目次


📍 クイックナビゲーション
14q近位欠失症候群は、第14番染色体長腕の動原体側(14q11〜14q22)に位置する微小な欠失によって生じる、極めて稀少な染色体異常症のスペクトラムです。欠失する位置によって全く異なる病態を示し、14q11.2欠失では大頭症と自閉症的特徴、14q12欠失では脳梁欠損と重度神経退行(FOXG1症候群)、14q13欠失では先天性甲状腺機能低下症と新生児呼吸窮迫(脳肺甲状腺症候群)など、責任遺伝子ごとに固有の臨床像を呈します。
かつてGバンド染色体検査の時代には「14q近位欠失症候群」として一括りに扱われていましたが、染色体マイクロアレイ検査(CMA)やCNV-seqの普及により、それぞれが固有の責任遺伝子と表現型を持つ独立した微小欠失症候群であることが解明されました。CHD8・FOXG1・NKX2-1などの転写制御因子のハプロ不全が病態の中心にあります。
本記事では、14q近位欠失症候群を構成する各サブタイプの病態・症状・診断・治療・予後について、最新の分子遺伝学的知見と臨床データをもとに、臨床遺伝専門医の視点から網羅的に解説します。
1. 14q近位欠失症候群とは|疾患の基本情報
14q近位欠失症候群は、第14番染色体長腕のうち動原体(セントロメア)に比較的近い領域(おおむね14q11〜14q22のバンド帯)で起こる染色体微小欠失の総称です。第14番染色体は端部着糸型(アクロセントリック)染色体に分類され、短腕(14p)にはタンパク質コード遺伝子は含まれていないため、臨床的に意味を持つ欠失はほぼすべて長腕(14q)で起こります。
1970〜1990年代のGバンド法による核型分析では微小な欠失の境界を正確に同定することが困難で、様々な欠失サイズの患者がすべて「14q近位欠失症候群」として一括して報告されていました。しかし、染色体マイクロアレイ検査(CMA)・アレイCGH・CNV-seqなどの分子細胞遺伝学的手法の普及により、それぞれが固有の責任遺伝子と表現型を持つ独立した微小欠失症候群であることが解明されました。
染色体上の非常に小さな領域(数キロベース〜数メガベース)が失われることで起こる症候群の総称です。光学顕微鏡で見るGバンド法では検出できないほど小さいため、染色体マイクロアレイ検査(CMA)や次世代シーケンシングといった高解像度の分子検査が必須となります。22q11.2欠失症候群やプラダー・ウィリ症候群なども、このグループに含まれます。
1.1 14q近位領域に存在する主な微小欠失症候群
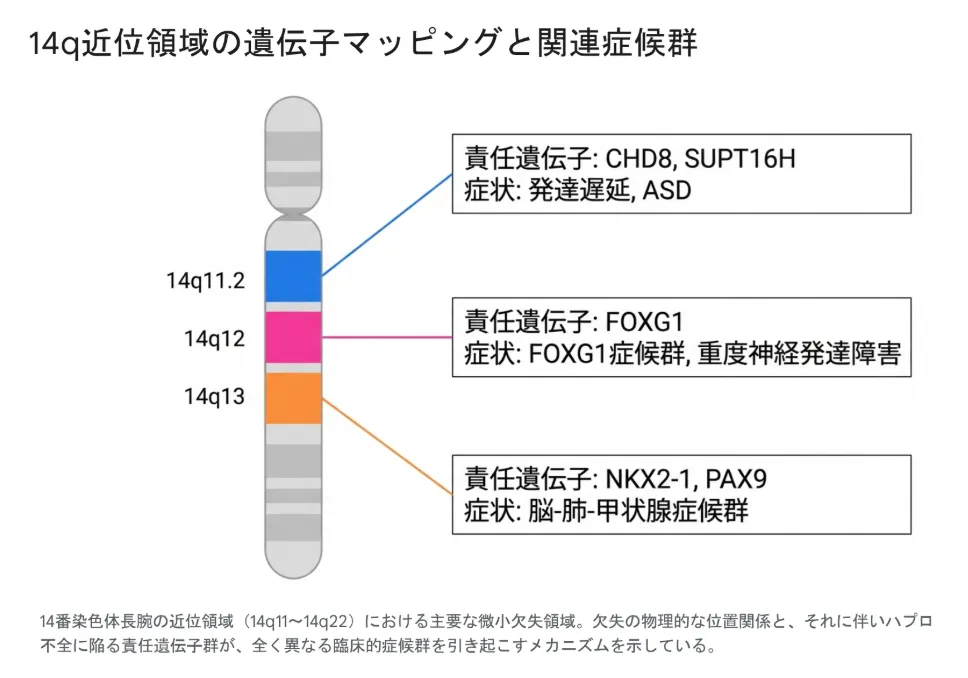
14q近位領域には複数のマスター制御遺伝子が密に並んでおり、欠失する位置によって全く異なる症候群を引き起こします。
| 領域 | 主な責任遺伝子 | 代表的な疾患・症状 |
|---|---|---|
| 14q11.2 | CHD8、SUPT16H | 14q11.2微小欠失症候群(発達遅滞・大頭症・ASD様症状) |
| 14q12 | FOXG1 | FOXG1症候群(後天性小頭症・脳梁欠損・重度神経退行) |
| 14q13 | NKX2-1、PAX9 | 脳肺甲状腺症候群(BLTS)(舞踏病・先天性甲状腺機能低下症・呼吸窮迫) |
| 14q11-q22 | 複数遺伝子の同時喪失 | 隣接遺伝子症候群(重症・多臓器障害) |
1.2 共通する基本情報
| 項目 | 内容 |
|---|---|
| 疾患名 | 14q近位欠失症候群(Proximal 14q deletion syndrome) |
| 頻度 | 14q11.2微小欠失単独では100万人に1人未満。FOXG1症候群・BLTSもそれぞれ希少 |
| 遺伝形式 | 大半が新生突然変異(de novo)。常染色体顕性(優性)形式で稀に遺伝。生殖細胞モザイクによる反復例も報告 |
| 診断方法 | 染色体マイクロアレイ検査(CMA)が標準。WES/CNV-seqも有用 |
| 治療 | 根本治療なし。症状に応じた多職種チーム医療が中心 |
2. 14q11.2微小欠失症候群(CHD8/SUPT16H関連)
14q11.2領域の限局的な微小欠失は、広汎な発達遅滞・全般的な筋緊張低下・特有の顔貌異常を三主徴とする独立した症候群です。発生頻度は100万人に1人未満と極めて稀少ですが、CMAの普及により診断例は増えつつあります。
2.1 責任遺伝子|最小共通欠失領域は35kb
高解像度のオリゴヌクレオチドアレイ解析により、異なる民族的背景を持つ複数の患者間で、表現型を決定づける最小共通欠失領域(Minimal Critical Region)がわずか35キロベース(kb)にまで絞り込まれています。この極めて狭小な領域には、2つの主要な遺伝子が含まれています。
- CHD8:クロマチンリモデリング因子をコードし、中枢神経系の発生において転写調節の中心的役割を担う。自閉症スペクトラム障害(ASD)の最も強力な単一原因遺伝子の一つ。大頭症・自閉症的行動の主因と考えられている。
- SUPT16H:転写伸長を促進するヒストンシャペロン複合体FACTのサブユニット。認知機能障害・運動発達遅滞に相加的に寄与。
- HNRNPC:欠失領域直近に位置し、「位置効果」により発達遅滞の増悪に関与する可能性。
- DAD1:一部の患者で見られる合指症(第2趾と第3趾の皮膚性癒合)に関与。
通常、私たちの遺伝子は父と母から1コピーずつ、計2コピー受け継いでいます。片方のコピーが欠失または機能しなくなることで、残った1コピーだけでは正常な機能を維持できない状態を「ハプロ不全」と呼びます。14q近位欠失症候群では、CHD8・FOXG1・NKX2-1などの転写制御因子のハプロ不全が病態の中心となります。
2.2 神経発達と行動学的プロファイル
幼児期から運動機能(這い這い・自立歩行)および言語発達の顕著な遅れを呈します。多くの場合、軽度から中等度の知的障害が認められ、特に表出言語が損なわれる傾向があります。
- ADHD様症状・衝動性:約40%
- 攻撃的行動:約53%
- 自閉症スペクトラム障害(ASD)診断基準を満たす患者:約36%(エコラリア・常同運動・こだわり)
- てんかん:約27%(欠神発作や点頭てんかん。多くは抗てんかん薬で管理可能)
- 肥満・過食症:約47%
2.3 頭蓋顔面の特徴|大頭症が決定的サイン
14q11.2欠失で最も特徴的なのが、CHD8のハプロ不全に起因する大頭症(頭囲が97パーセンタイル以上)・巨脳症です。これは後述する14q12欠失が小頭症を呈する点と決定的に異なる鑑別ポイントとなります。
- 眼間開離(両目の間隔が広い)
- 平坦で幅の広い鼻梁、短鼻
- 深い・長い人中、顕著なキューピッドの弓
- 下唇の肥厚・外反、口角の下降
- 高くアーチ状の眉、後方回転を伴う低位耳
3. 14q12欠失とFOXG1症候群|脳発生の根幹を揺るがす疾患
14q11.2からさらに遠位の14q12領域の欠失は、極めて重篤な神経発達障害を引き起こします。この領域の中心的な責任遺伝子はFOXG1(Forkhead box G1)で、その欠失や変異はレット症候群の先天性バリアントとも酷似した独立した疾患概念「FOXG1症候群」を形成します。
3.1 FOXG1の神経生物学的機能
FOXG1は、発達中の脳において特異的に発現するフォークヘッドドメイン含有転写因子をコードしています。胎生期の動物モデル研究では、FOXG1を過剰発現させると神経上皮細胞のアポトーシスが減少し、神経上皮の肥厚と終脳・中脳の著しい成長が誘導されることが実証されています。逆に、FOXG1のハプロ不全は神経前駆細胞の過剰なアポトーシスと増殖不全を引き起こし、脳の容積低下を招きます。
注目すべき分子遺伝学的発見として、FOXG1遺伝子のコーディング領域そのものが無傷であっても、下流の制御領域(長距離エンハンサー)のみが欠失することで発現量が著しくダウンレギュレートされ、典型的なFOXG1症候群を呈する症例が複数報告されています。3次元的なクロマチン構造と遠隔転写調節の破綻が疾患発症に重要であることを示す重要な知見です。
3.2 臨床的軌跡|出生時正常→生後3〜6ヶ月で退行
FOXG1症候群の臨床的軌跡は非常に特徴的です。出生直後の新生児期は一見正常に経過することが多いものの、生後3〜6ヶ月の間に急速な発達の退行を迎えます。この退行期に頭囲の成長が著しく減速し、結果として重度の「後天性小頭症」へと至ります。
- 後天性小頭症:出生時正常→生後数ヶ月で頭囲発育停滞
- 脳梁欠損・低形成:ほぼ全例で確認される画像的ホールマーク
- 自立歩行・有意義な言語獲得:大部分で到達せず
- 筋緊張の不均衡:四肢に進行性痙縮、体幹は低緊張
- 難治性てんかん:乳児スパスム・欠神発作。発達性てんかん性脳症の様相を呈することも
- 常同運動:レット症候群類似の「手のもみ動作」、口や舌の異常運動
- 重度睡眠障害:家族のQOLを著しく損なう要因
4. 14q13欠失と脳肺甲状腺症候群(BLTS)
14q12からさらに遠位の14q13領域、特に14q13.2〜14q13.3の微小欠失は、中枢神経系のみならず呼吸器系および内分泌系の複合的な臓器形成不全をもたらします。
4.1 中核遺伝子NKX2-1(旧TITF-1)と三徴候
中核的な責任遺伝子は転写因子NKX2-1(旧称:TITF-1 / Thyroid Transcription Factor-1)です。胎生期に脳の基底核・肺の上皮細胞・甲状腺の原基に強く発現し、これらの臓器の発生と分化に必須の役割を果たします。NKX2-1のハプロ不全は「脳・肺・甲状腺症候群(Brain-Lung-Thyroid Syndrome: BLTS)」を引き起こします。
①神経学的異常:良性遺伝性舞踏病(BHC)を筆頭に、舞踏アテトーゼ・振戦・ジストニア・運動と音声チック等の不随意運動が幼少期〜思春期に発症。
②先天性甲状腺機能低下症:甲状腺の発生異常により新生児期から傾眠・便秘・筋緊張低下・巨舌・遷延性黄疸として発現。
③呼吸器系疾患:サーファクタント産生制御の異常により出生直後から重篤な新生児呼吸窮迫症候群(RDS)、小児期の間質性肺疾患(ILD)、年長児の肺線維症。致死的呼吸不全のリスクあり。
表現型の浸透率は多様で、患者の約50%が完全な三徴候、約30%が脳と甲状腺のみ、約13%が舞踏病のみを呈すると報告されています。
4.2 PAX9遺伝子と歯科的表現型
欠失領域がNKX2-1近傍のPAX9遺伝子にまで及ぶ場合、PAX9のハプロ不全に起因して「多数歯欠損(オリゴドンチア)」または「無歯症」という明確な歯科的表現型が付加されます。
4.3 ホロプロスエンセファリー(HPE8)遺伝子座
14q13領域にはホロプロスエンセファリー(前脳胞分芽不全:HPE)の関連遺伝子座「HPE8」が存在することが確認されています。研究者らは2.82 Mbの最小限界領域を特定し、胎児脳で発現するNPAS3・SNX6・C14ORF11などの候補遺伝子を同定しています。14q13欠失が基底核や肺の発生にとどまらず、脳の初期の全体構造のパターニングにも影響する可能性を示唆します。
5. 広範な近位間質性欠失(14q11-q22)|隣接遺伝子症候群
14q11から14q22にかけて数メガベースから十数メガベースにわたって広範囲に欠失するケースでは、複数の重要な遺伝子が同時に失われる「隣接遺伝子症候群(contiguous gene syndrome)」の形態をとり、各領域の症状が重層的に現れます。表現型は極めて複雑かつ重篤になります。
- 14q13と14q21を含む欠失:NKX2-1欠失による重篤な新生児呼吸不全に加え、極度の発育不全(Failure to thrive)。重度の胃食道逆流症(GERD)が頻発し、反復性の誤嚥性肺炎に苦しむ症例も。ニッセン噴門形成術や胃瘻造設を要する場合がある。
- 心血管系の奇形:心室中隔欠損症(VSD)・動脈管開存症(PDA)・肺動脈分岐部狭窄など。血行動態的負荷が呼吸器合併症と相まって生命予後を悪化させる。
- 骨格異常:低身長、第5中手骨・中足骨の短縮、関節拘縮など。
6. 領域別の臨床所見比較|遺伝子型と表現型の相関
14q11.2・14q12・14q13の各微小欠失領域は、症状の中核要素が全く異なります。以下の比較表は、それぞれの「決定的特徴」「共通特徴」「非典型的所見」を視覚的に整理したものです。
📊 14q近位欠失における領域別の主要臨床所見
共通特徴(Medium)
非典型的(Low)
| 身体系統 / 症状 | 14q11.2 (CHD8/SUPT16H) |
14q12 (FOXG1) |
14q13 (NKX2-1) |
|---|---|---|---|
| 神経・発達 発達遅滞 / 知的障害 |
+++ 重度遅滞 |
+++ 重度・退行 |
++ 軽度〜重度 |
| 頭部形状 大頭症 / 小頭症 |
++ 大頭症 |
+++ 生後小頭症 |
− 非典型的 |
| 神経系 てんかん・発作 |
++ 一部にみられる |
++ 共通 |
− 非典型的 |
| 運動障害 舞踏運動(Chorea) |
− 非典型的 |
− 非典型的 |
+++ 舞踏アテトーゼ |
| 内分泌系 甲状腺機能低下症 |
− 非典型的 |
− 非典型的 |
+++ 先天性低下症 |
| 呼吸器系 肺疾患・呼吸窮迫 |
− 非典型的 |
− 非典型的 |
+++ RDS / 肺機能不全 |
| 脳構造 脳梁欠損・低形成 |
− 比較的保たれる |
+++ 高頻度に欠損 |
− 非典型的 |
| 身体的特徴 特異的顔貌 |
+++ 顕著 |
++ 共通 |
− 孤発的 |
14q11.2、14q12、14q13の各微小欠失領域に関連する主要な臨床的特徴の比較。濃い青色は決定的特徴、水色は共通特徴、グレーは関連性が低いか報告が少ない特徴を示しています。
7. 診断方法と鑑別診断
14q近位欠失症候群の診断において、従来のGバンド法は微小欠失の検出に限界があります。正確な確定診断と、遺伝子型-表現型相関に基づく予後予測のためには、高解像度の分子細胞遺伝学的検査が必要です。
7.1 検査方法ごとの違い
| 検査方法 | 特徴 | 14q近位欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 確定診断のゴールドスタンダード。数kb単位の欠失も検出 | ◎ 確実に検出(35kb欠失も特定可能) |
| CNV-seq | 次世代シーケンシングによるCNV解析 | ◎ 高解像度で検出可能 |
| 全エクソームシーケンス(WES) | 遺伝子の塩基配列を網羅的に解析 | ○ 解析設定により可能。点突然変異も同時検出 |
| Gバンド法(核型分析) | 解像度は約5〜10Mb | ✕ 検出困難(微小欠失は見逃される) |
| FISH法 | 特定領域のプローブで視覚的確認 | △ 専用プローブで可能 |
CMA(chromosomal microarray analysis)は、従来のGバンド法では検出できない数kb〜数Mb単位の微小な欠失や重複(コピー数変異:CNV)を網羅的に検出する検査です。日本では原因不明の発達遅滞・知的障害・多発奇形に対する保険適用検査として実施されており、14q近位欠失症候群の確定診断には欠かせません。
7.2 鑑別診断|FOXG1症候群と紛らわしい疾患
特にFOXG1症候群はレット症候群(MECP2変異)と臨床像が酷似しており、両者の鑑別が重要です。後天性小頭症・常同運動(手のもみ動作)・自閉症的特徴・てんかんなど共通点が多いため、CMAとMECP2解析を同時に行うことで適切な確定診断が可能です。
7.3 生殖細胞モザイクへの注意
14q近位欠失の大部分は新生突然変異として発生しますが、臨床現場で慎重な対応を要するのが「生殖細胞系列モザイク(Gonadal mosaicism)」です。親自身の末梢血染色体検査が正常であっても、生殖細胞(精子や卵子)の一定割合にのみ14q欠失が存在する場合、健康な親から同じ欠失を持つ子どもが反復して生まれるリスクがあります。実際に、親のモザイクに起因して、14q13.3欠失による重度の無歯症・先天性甲状腺機能低下症・肺高血圧症を呈する兄弟(Siblings)の症例が文献で報告されています。
8. 治療と長期管理|多職種チームによる包括的サポート
14q近位欠失症候群には根本的な治療法はまだ存在しません。治療は症状に応じた対症療法・早期療育・継続的支援が中心となり、小児科を司令塔とした多職種チーム医療が不可欠です。
8.1 主な医療分野と対応
| 医療分野 | 対象となる主な症状 | 推奨される治療介入 |
|---|---|---|
| 小児神経・小児科 | てんかん(特に14q12)、睡眠障害 | 抗てんかん薬の調整、脳波モニタリング |
| 神経・運動器 | 舞踏病・アテトーゼ(NKX2-1) | テトラベナジン・デュテトラベナジン等の低用量投与、レボドパ |
| リハビリテーション | 痙縮、筋緊張低下、発達遅滞 | PT/OT、バクロフェン、ボツリヌス毒素の局所投与 |
| 内分泌科 | 先天性甲状腺機能低下症(NKX2-1) | レボチロキシン(LT4)補充療法。新生児期の早期開始が脳保護に重要 |
| 呼吸器科 | RDS、間質性肺疾患、肺癌リスク | 新生児期の人工呼吸サポート、RSVワクチン、若年成人期の肺癌スクリーニング |
| 消化器・外科 | 嚥下障害、GERD、重度便秘 | 緩下剤・消化管運動薬、胃瘻造設、ニッセン噴門形成術 |
| 心理社会的支援 | 家族介護負担、教育リソース | レスパイトケア、特別支援教育計画(IEP)、患者支援団体 |
8.2 早期介入の重要性
理学療法・作業療法・言語療法(AAC:拡張代替コミュニケーションを含む)・視覚療法への乳幼児期からの早期アクセスは、神経回路の可塑性が高い時期に介入することで、お子さんの潜在的な能力を最大限に引き出すための鍵となります。BLTS(14q13欠失)の場合は、生後早期の甲状腺ホルモン補充が脳への不可逆的ダメージを防ぐ上で決定的に重要です。
8.3 長期予後|サブタイプ別の生存率
- FOXG1症候群(14q12):病理学的には進行性神経変性ではなく「安定した脳症」。退行は乳児期に限定され、その後の継続的なスキル喪失は稀。生命予後を左右するのは難治性てんかんの合併症や誤嚥性肺炎で、適切な医療的ケアにより32歳の成人期まで生存している症例も報告されています。
- 脳肺甲状腺症候群(14q13):呼吸器症状の重症度が生命予後を決定。文献のシステマティックレビューでは全生存率が約60%と推定されています。若年成人期以降の肺癌リスクにも注意が必要。
- 14q11.2微小欠失:心血管系の重大な奇形や呼吸器障害を伴わない限り、生命予後は比較的良好。14歳までの長期フォロー報告でも安定した生存と成長が確認されています。
- 広範な14q11-q22欠失:複数の致死的要因が重なるため予後不良。生後数ヶ月以内の死亡例も過去に報告。
9. 遺伝カウンセリングと出生前診断|ミネルバクリニックのサポート
14q近位欠失症候群はサブタイプによって予後が大きく異なり、表現型の幅も広い疾患です。遺伝カウンセリングを通じて、ご家族が病気を正確に理解し、納得のいく決断ができるよう中立的な情報提供を行うことが、医師の重要な役割です。
9.1 出生前検査と14q近位欠失
ミネルバクリニックのNIPTのうち、ターゲット法による高精度検査であるダイヤモンドプランでは特定12箇所の微小欠失(1p36、4p16、5p15、9p、22q11.2など)を対象としますが、14q近位欠失はこの12箇所には含まれません。一方、インペリアルプランはWGS法とターゲット法のハイブリッドで、5Mb以上の全染色体微小欠失・重複を広範囲にスクリーニングするため、広範な14q近位欠失もカバー対象となります。
| 検査 | 位置づけ | 14q近位欠失への対応 |
|---|---|---|
| NIPT(ターゲット型:12箇所) | スクリーニング検査 | ✕ 対象外(14qは12微小欠失に含まれず) |
| NIPT(WGS型 / インペリアル) | スクリーニング検査 | ○ 5Mb以上の欠失を広くスクリーニング |
| 絨毛検査+CMA | 確定診断 | ◎ 妊娠初期に微小欠失も確定診断可能 |
| 羊水検査+CMA | 確定診断 | ◎ Gバンドでは検出困難な微小欠失も確定診断 |
⚖️ 倫理的なスタンス|検査は「常に利益」ではない
14q近位欠失症候群のようにサブタイプによって予後が大きく異なる疾患では、出生前に見つけたことが必ずしもご家族の利益になるとは限りません。「特定の検査を勧める」「安心を保証する」「不安をあおる」表現は適切ではないと私たちは考えています。検査を受けるかどうか、結果をどう受け止めるかは、十分な情報を得たうえで、ご家族自身が決めるべき事柄です。
9.2 ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医による診療体制を整えています。14q近位欠失症候群を含む染色体微小欠失症候群について、出生前検査から結果説明、確定検査、その後のフォローまで一貫してサポートいたします。
- 全染色体スクリーニング対応:インペリアルプランでは5Mb以上の全染色体微小欠失・重複を広くスクリーニング
- 確定検査も院内で実施:羊水検査・絨毛検査も院内で実施可能、転院の必要なし
- 臨床遺伝専門医が担当:検査前後の遺伝カウンセリングを1.5時間の枠をお取りして直接担当
- 互助会で費用面も安心:NIPT受検者全員に適用される互助会(8,000円)により、陽性時の羊水検査費用が全額補助されます
お子さんの発達や検査結果が気になっていませんか?
原因不明の発達遅滞や多発奇形には染色体マイクロアレイ検査が有効です。
臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
🧬 その他の染色体異常について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
よくある質問(FAQ)
関連記事
参考文献
- Orphanet – 14q11.2 microdeletion syndrome [外部サイトへ]
- Orphanet – FOXG1 syndrome due to 14q12 microdeletion [外部サイトへ]
- GeneReviews – FOXG1 Syndrome (NCBI Bookshelf) [外部サイトへ]
- GeneReviews – NKX2-1-Related Disorders (NCBI Bookshelf) [外部サイトへ]
- GARD (NIH) – Chromosome 14q11-q22 deletion syndrome [外部サイトへ]
- Zahir F et al. Novel deletions of 14q11.2 associated with developmental delay, cognitive impairment and similar minor anomalies. PMC. [外部サイトへ]
- Prontera P et al. Long term follow-up in a patient with a de novo microdeletion of 14q11.2 involving CHD8. PubMed PMID: 25735987 [外部サイトへ]
- Allou L et al. Dysregulation of FOXG1 pathway in a 14q12 microdeletion case. PubMed PMID: 23956198 [外部サイトへ]
- Epilepsy and outcome in FOXG1-related disorders. PMC. [外部サイトへ]
- Haploinsufficiency of NKX2-1 is likely to contribute to developmental delay involving 14q13 microdeletions. PMC. [外部サイトへ]
- Systematic review of thyroid function in NKX2-1-related disorders. PMC. [外部サイトへ]
- Respiratory and other organ manifestations in NKX2-1-related disorders: a systematic review. Frontiers in Medicine. [外部サイトへ]
- 14q13 distal microdeletion encompassing NKX2-1 and PAX9. PubMed PMID: 27148860 [外部サイトへ]
- Identical deletion at 14q13.3 including PAX9 and NKX2-1 in siblings from mosaicism. PubMed PMID: 25608831 [外部サイトへ]
- Defining a Holoprosencephaly Locus on Human Chromosome 14q13. PubMed PMID: 15820313 [外部サイトへ]
- The proximal chromosome 14q microdeletion syndrome (SOMA analysis). PubMed PMID: 21744488 [外部サイトへ]
- Rarechromo.org – 14q11.2 deletions FTNW [外部サイトへ]
- Rarechromo.org – 14q12 deletions FTNW [外部サイトへ]
- Rarechromo.org – 14q13 deletions FTNW [外部サイトへ]
- Rarechromo.org – 14q deletions proximal to 14q22 FTNW [外部サイトへ]






