目次
4p16欠失症候群(ウォルフ・ヒルシュホーン症候群)とは?
症状・原因・診断・治療を臨床遺伝専門医が解説
Q. 4p16欠失症候群(ウォルフ・ヒルシュホーン症候群)とはどのような病気ですか?
A. 第4番染色体短腕(4p16.3)の末端領域が欠失することで生じる、複雑な多系統疾患です。
「ギリシャ兵士のヘルメット様顔貌」と呼ばれる特徴的な顔つき、重度の成長遅滞、知的障害、難治性てんかんを主な特徴とします。出生頻度は約2〜5万人に1人の希少疾患です。
-
➤
原因 → 4番染色体短腕16.3領域(4p16.3)の欠失 -
➤
主要症状 → 特徴的顔貌(100%)、成長遅滞(100%)、知的障害(100%)、てんかん(90〜100%) -
➤
責任遺伝子 → NSD2・LETM1・MSX1などのハプロ不全 -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
頻度 → 出生約2〜5万人に1人、女性が男性の約2倍
1. 4p16欠失症候群(ウォルフ・ヒルシュホーン症候群)とは|基本情報
【結論】 4p16欠失症候群(Wolf-Hirschhorn Syndrome: WHS)は、第4番染色体短腕の末端領域(4p16.3)が欠失することで生じる希少な遺伝性疾患です。1960年代にWolf氏とHirschhorn氏によって報告されたことから「ウォルフ・ヒルシュホーン症候群」と呼ばれ、日本では指定難病198に指定されています。
「お子さんが4p16欠失症候群と診断された」「NIPTで4p欠失の疑いを指摘された」という方は、この病気について正確な情報を知ることが大切です。本症候群は連続遺伝子欠失症候群であり、欠失した領域の大きさによって症状の重さが異なります。
💡 用語解説:「連続遺伝子欠失症候群」とは?
染色体の一部が欠失することで、その領域に含まれる複数の遺伝子がまとめて失われる疾患群のことです。単一遺伝子疾患と異なり、欠失サイズによって失われる遺伝子の数が変わるため、症状のスペクトラム(軽症〜重症)が生じます。
4p16欠失症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 4p16欠失症候群(OMIM #194190) |
| 別名 | ウォルフ・ヒルシュホーン症候群(WHS)、4p-症候群 |
| 原因 | 4番染色体短腕16.3領域(4p16.3)の欠失 |
| 頻度 | 出生約2〜5万人に1人(日本では推定1,000人以下) |
| 性差 | 女性:男性 ≒ 2:1 |
| 遺伝形式 | 85〜90%が新生突然変異(de novo)、10〜15%が親の均衡型転座由来 |
| 責任遺伝子 | NSD2(WHSC1)、LETM1、MSX1など |
⚠️ WHS核心領域(WHSCR)について
WHSの責任領域は、4p末端から約1.4〜1.9Mbに位置する約165kbの極めて狭い「WHS核心領域(WHSCR)」に絞り込まれています。この領域を含む欠失がWHSの核心的な症状(特徴的顔貌・成長障害)を引き起こします。欠失がより大きい場合は、追加の症状が現れます。
なぜ女性に多いのか?
WHSでは女性の発症率が男性の約2倍という顕著な性別バイアスがあります。その理由として以下の仮説が挙げられていますが、決定的な結論には至っていません。
仮説①:胚発生時の生存率の差
男性胚は4p欠失があると胎児期に死亡しやすい可能性。結果として出生する患者は女性が多くなる。
仮説②:X染色体との相互作用
4番染色体上の遺伝子とX染色体の遺伝子の相互作用により、女性(XX)の方が保護効果を受けている可能性。
2. 4p16欠失症候群の主な症状
【結論】 本症候群は特徴的な顔貌(「ギリシャ兵士のヘルメット様」)、重度の成長遅滞、知的障害、てんかん(90〜100%)を主な特徴とします。加えて、心奇形、骨格異常、腎泌尿器異常、免疫不全など多臓器にわたる合併症を伴うことがあります。
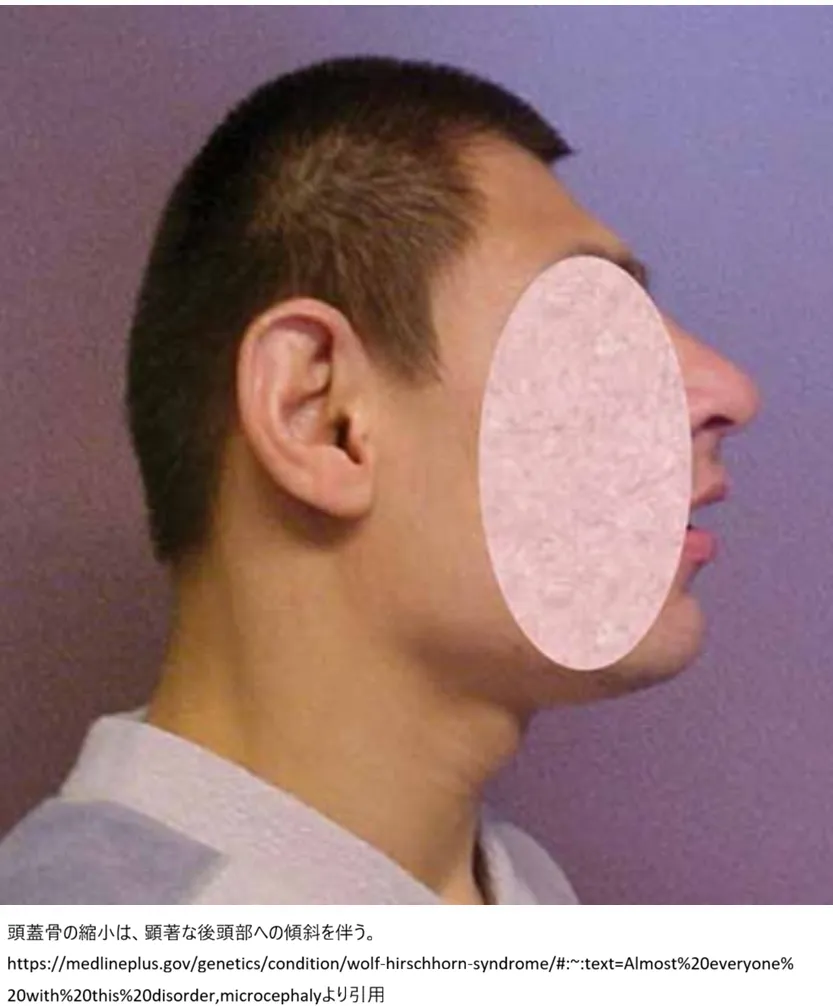
特徴的顔貌:「ギリシャ兵士のヘルメット様顔貌」
WHSを診断する上で最も直感的かつ決定的な指標は、独特の顔立ちです。古代ギリシャの兵士が着用した鼻当て付きのヘルメットを想起させる構造から名付けられました。
-
•
鼻梁・前頭部:非常に幅広く平坦な鼻梁が高い前頭部へと連続
-
•
眉間・眼部:眉間が突出、眼間開離(両眼の間隔が広い)、眼球突出傾向
-
•
口部・顎部:短い人中、口角下垂(「への字型」の口)、極めて小さな下顎(小顎症)
-
•
耳介:耳の形成不全、耳前瘻孔(耳の前の小さな穴)、副耳
症状の出現頻度一覧
| 症状カテゴリー | 頻度 | 詳細 |
|---|---|---|
| 特徴的顔貌 | 100% | 「ギリシャ兵士のヘルメット様顔貌」 |
| 成長遅滞 | 100% | 出生前からの発育不全、低身長、体重増加不良 |
| 知的障害 | 100% | 中等度〜重度(IQ 35未満が多い)、言語表出が特に困難 |
| てんかん | 90〜100% | 生後3〜36ヶ月に発症、発熱が誘因となりやすい |
| 筋緊張低下 | 高頻度 | 全身性の筋力低下、運動発達遅延 |
| 先天性心疾患 | 約50% | 心房中隔欠損(ASD)、心室中隔欠損(VSD)など |
| 骨格異常 | 60〜70% | 脊柱側弯症、後弯症、内反足、肋骨異常 |
| 泌尿生殖器異常 | 25〜50% | 腎低形成、嚢胞腎、尿道下裂、停留精巣 |
| 免疫不全 | 高頻度 | IgA・IgG2欠損、繰り返す呼吸器感染症 |
| 口唇口蓋裂 | 一部 | MSX1遺伝子欠失に関連 |
てんかん:管理上の最大の課題
WHS患者の約90〜100%に認められるてんかんは、生活の質および生命予後を左右する重要な要素です。
-
•
発症時期:生後3〜36ヶ月に初発が多い
-
•
誘因:発熱が強力な誘発因子、熱性けいれん重積状態も
-
•
発作型:全般性強直間代発作、非定型欠神発作、ミオクロニー発作など多様
-
•
長期経過:乳幼児期は難治性だが、思春期までに約半数で発作消失
💡 行動表現型:社交的な性格
知的障害は重度であることが多いですが、WHS患者には「陽気で社交的な性格」という特徴的な行動表現型が知られています。受容的なコミュニケーションや社会性、他者への関心は比較的保たれていることが多く、これは家族にとって大きな喜びとなります。

🩺 院長コラム【WHSの「表現型の多様性」について】
4p16欠失症候群を診療していると、同じ「WHS」でも症状の幅がとても広いことに気づきます。これは「連続遺伝子欠失症候群」の特徴であり、欠失サイズが大きいほど重症になる傾向があります。
また、10〜15%の症例では親の均衡型転座に由来しますが、この場合は4p欠失だけでなく他の染色体の部分重複を伴うことがあり、症状がより複雑になります。
大切なのは、「WHSだから○○」と一括りにしないことです。一人ひとりの欠失範囲を正確に評価し、その方に合った管理計画を立てることが重要です。
3. 原因と遺伝的背景|3つの責任遺伝子
【結論】 WHSの病因は4p16.3領域の欠失です。85〜90%は新生突然変異で偶発的に生じますが、10〜15%は親の均衡型転座に由来します。責任遺伝子としてNSD2(顔貌・成長)、LETM1(てんかん)、MSX1(口蓋裂・歯)が同定されています。
欠失の発生パターン
| 欠失のタイプ | 頻度 | 遺伝学的特徴 |
|---|---|---|
| 新生突然変異(de novo) | 85〜90% | 両親の染色体は正常。精子・卵子形成時または胚発生初期に偶発的に生じる |
| 不均衡型転座 | 10〜15% | 親が均衡型転座の保因者。4p欠失+他染色体の部分重複を伴うことあり |
| 環状染色体(Ring 4) | 稀 | 4番染色体両端が切断され環状に結合、末端遺伝子が消失 |
💡 用語解説:「均衡型転座」とは?
染色体の一部が別の染色体と入れ替わった状態です。保因者自身は遺伝物質の過不足がないため健康ですが、配偶子(精子・卵子)形成時に不均衡な分配が起こると、子どもに欠失や重複が生じることがあります。
3つの主要責任遺伝子
| 遺伝子 | 主な機能 | 関連する臨床症状 |
|---|---|---|
| NSD2(WHSC1) | ヒストンメチルトランスフェラーゼ、クロマチン制御 | 特徴的顔貌、成長障害(WHSの核心的症状) |
| LETM1 | ミトコンドリアCa²⁺/H⁺アンチポーター | てんかん(脳内電気活動異常) |
| MSX1 | 神経堤細胞分化、形態形成の転写抑制因子 | 口唇口蓋裂、歯の欠損、性分化疾患(DSD) |
-
①
エピジェネティック制御:ヒストンH3K36のメチル化を担い、多数の遺伝子発現を制御
-
②
DNA損傷応答:NSD2欠乏はDNA修復不全を引き起こし、神経発達への影響も示唆
-
③
核心的役割:NSD2のハプロ不全がWHSの顔貌と成長障害の主因
⚠️ 欠失サイズと重症度の関係:WHSでは一般に欠失サイズが大きいほど症状が重症化します。核心領域(約165kb)のみの欠失では軽症型、数Mbに及ぶ大きな欠失では多臓器合併症を伴う重症型となります。
4. 4p16欠失症候群の診断方法
【結論】 WHSの確定診断には染色体マイクロアレイ検査(CMA)が第一選択です。従来のG分染法では約42%の症例を見逃す可能性があり、臨床的に疑わしい場合はCMAまたはFISH法による精査が必須です。
診断のきっかけ
WHSは特徴的な臨床所見から疑われ、遺伝学的検査で確定します。
-
①
特徴的顔貌:「ギリシャ兵士のヘルメット様」の顔つき
-
②
重度の発育不全:子宮内胎児発育不全(IUGR)、低出生体重、発育不良
-
③
乳児期てんかん:特に発熱で誘発される難治性けいれん
-
④
複合所見:上記+心奇形、口蓋裂、骨格異常など
遺伝学的検査の種類
| 検査方法 | 特徴 | WHS検出能 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 第一選択・ゴールドスタンダード。欠失サイズを正確にマッピング | ◎ 高精度で検出可能 |
| FISH法 | WHSCR領域プローブで迅速に確認。転座保因者検査にも有用 | ◎ 検出可能 |
| G分染法(核型分析) | 伝統的方法。大きな転座や数的異常を検出 | △ 約42%を見逃す可能性 |
⚠️ G分染法の限界
G分染法の解像度は5〜10Mb程度であり、WHSの原因となる微小欠失(数百kb〜数Mb)の約42%を検出できないことが報告されています。G分染法が陰性でもWHSを否定することはできません。臨床的に疑われる場合はCMAまたはFISH法での精査が必須です。
鑑別診断
WHSは多岐にわたる症状を呈するため、以下の疾患との鑑別が重要です。
アンジェルマン症候群/レット症候群
知的障害とてんかんが主徴だが、WHSに特有の顔貌や心奇形は通常見られない
CHARGE症候群
コロボーマや心奇形を伴うが、原因遺伝子(CHD7)が異なり、顔貌も識別可能
Seckel症候群
小頭症と成長障害を伴うが、WHSCR領域の欠失は認められない
5. 治療と長期管理
【結論】 WHSには根本的な治療法は存在せず、症状に応じた対症療法・早期療育・多職種連携による包括的ケアが中心となります。てんかん管理、栄養サポート、リハビリテーションが特に重要です。
治療の柱
⚡ てんかん治療
- •
第一選択:バルプロ酸
- •
非定型欠神発作にはエトスクシミド併用
- •
レベチラセタムも使用報告あり
- •
カルバマゼピンは回避(発作悪化リスク)
🍽️ 栄養・摂食支援
- •
筋緊張低下による哺乳困難への対応
- •
必要に応じて経管栄養・胃瘻(PEG)
- •
胃食道逆流症(GERD)管理で誤嚥性肺炎予防
🏃 リハビリテーション
- •
早期療育が最も重要
- •
理学療法(PT):筋力強化、移動支援
- •
作業療法(OT):日常生活動作獲得
- •
言語療法(ST):代替コミュニケーション手段構築
🏥 合併症の管理
- •
心奇形:定期心エコー、必要時外科的修復
- •
骨格異常:側弯症の定期評価、装具療法
- •
免疫不全:感染予防、予防接種
⚠️ 避けるべき事項
- カルバマゼピンの回避:脳波異常や非定型欠神発作を悪化させる恐れがあります
- 診断の遅れ:心奇形や腎奇形は早期発見が予後を左右します。臨床的疑いがある段階で全身スクリーニングを実施してください
最新研究(2024-2025年):治療の新展開
📊 GFAPスコア(2024-2025)
WHS患者の重症度を多角的に評価する新しい指標。発達マイルストン、てんかん制御状態、合併症を統合した機能評価スコアとして臨床試験での活用が期待されています。
💉 成長ホルモン療法(2025)
部分的成長ホルモン欠損を伴うWHS症例で、11年間のGH療法により身長SDSが-4.2から-1.3へ改善。筋緊張低下の改善も報告されました。
🧬 CRISPR遺伝子治療(将来展望)
連続遺伝子欠失のため単一遺伝子置換より複雑ですが、NSD2やLETM1をターゲットにした脂質ナノ粒子送達技術の研究が進行中です。
予後と生命予後
現代の支持療法の進歩により、WHSの生存率は大幅に改善しています。
📈 予後について
- 生存期間:多くの患者が成人期を迎え、30〜40代での生存報告も増加
- 乳幼児期の死因:重度心奇形、てんかん重積状態、繰り返す呼吸器感染症
- 成人期の自立度:約30%は日常のルーチンを理解し、補助付きでセルフケア可能
- てんかん:思春期までに約半数で発作が消失
6. 遺伝カウンセリングの重要性
【結論】 WHSの遺伝カウンセリングでは、欠失の発生機序(新生突然変異 vs 親の転座)を明らかにすることが再発リスク評価の鍵となります。親が均衡型転座の保因者である場合は、次子のリスクが高くなるため、着床前診断や出生前診断の選択肢について情報提供します。
再発リスクの評価
| 状況 | 次子への再発リスク | 備考 |
|---|---|---|
| 両親とも正常(新生突然変異) | 1%未満 | 生殖細胞モザイクの可能性は低いがゼロではない |
| 片親が均衡型転座の保因者 | 最大25%程度 | 転座の種類・分離パターンにより変動 |
-
①
両親の染色体検査:均衡型転座の有無を確認し、再発リスクを評価
-
②
欠失サイズと予後:欠失範囲と症状の関係についての情報提供
-
③
次子への選択肢:着床前診断・出生前診断の情報提供
-
④
心理的サポート:家族の不安・疑問に寄り添い、意思決定を支援
⚠️ 重要:転座由来の不均衡型転座の場合、母親由来の転座は父親由来の約2倍の頻度で伝達されることが知られています。両親の検査結果は、次子の遺伝カウンセリングにおいて極めて重要な情報となります。
🩺 院長コラム【WHSのご家族に寄り添って】
WHSの診断を受けたご家族にとって、最初に抱く疑問は「なぜうちの子に?」「私たちのせい?」ということです。85〜90%は偶発的に生じる新生突然変異であり、ご両親に原因があるわけではありません。
一方で、親が均衡型転座の保因者であった場合、次子への再発リスクを考慮する必要があります。この場合は着床前診断や出生前診断という選択肢があります。
また、WHSのお子さんは重度の障害を抱えながらも、「陽気で社交的」という魅力的な性格を持つことが多いです。その笑顔が家族の絆を深めている例をたくさん見てきました。
日本には「4p-症候群(WHS)親の会」などの患者家族団体もあります。同じ経験を持つ家族との交流は大きな支えになります。どんなことでもご相談ください。
7. 出生前診断について|NIPTと羊水検査
【結論】 4p16欠失症候群は出生前診断で検出可能です。ミネルバクリニックのNIPTでは4p16欠失を含む12種類の微小欠失症候群をスクリーニングできます。確定診断には羊水検査でのCMAが必要です。
出生前検査での検出方法
| 検査 | 特徴 | 備考 |
|---|---|---|
| NIPT(微小欠失検査) | スクリーニング検査 | ミネルバでは4p16欠失を含む12種の微小欠失に対応。COATE法採用で高精度 |
| 羊水検査+CMA | ◎ 確定診断 | Gバンド法では検出できない微小欠失を確定診断可能。 ※学会指針では、原則として超音波での構造異常がある場合などが対象とされています。 |
| 絨毛検査+CMA | ◎ 確定診断 | 妊娠初期(11〜14週)に実施可能 |
| 超音波検査 | スクリーニング | IUGR(胎児発育不全)、心奇形、口唇口蓋裂などを検出 |
💡 ミネルバクリニックのNIPTで検査可能な12種類の微小欠失
1p36欠失、2q33欠失、4p16欠失(本症候群)、5p15欠失、8q23q24欠失、9p欠失、11q23q25欠失、15q11.2-q13欠失、17p11.2欠失、18p欠失、18q22q23欠失、22q11.2欠失
出生前診断で見つかった場合
妊娠中の超音波検査で胎児発育不全(IUGR)や先天奇形(心疾患・口唇口蓋裂など)が見つかった場合、WHSが疑われることがあります。確定診断のためには羊水検査でのCMAが必要です。
-
①
遺伝カウンセリング:欠失の意味、予後、再発リスクを説明
-
②
両親の染色体検査:均衡型転座の有無を確認
-
③
詳細超音波:心奇形、口蓋裂など構造異常の精査
-
④
出生後フォロー体制:多職種チームによる管理計画
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。4p16欠失症候群を含む染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。4p16欠失を含む12種類の微小欠失症候群をスクリーニング可能です。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会制度で費用面も安心
互助会(8,000円)により、陽性時の確定検査(羊水検査)費用を全額補助。上限なしで安心です。
一人で悩まず、専門医を頼ってください
4p16欠失症候群について詳しく知りたい方、
出生前検査を検討している方は臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
🏥 一人で悩まないでください
4p16欠失症候群(ウォルフ・ヒルシュホーン症候群)について心配なこと、
検査を受けるかどうか迷っていること、どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
参考文献
- [1] Battaglia A, et al. Wolf-Hirschhorn Syndrome. GeneReviews. University of Washington, Seattle. [GeneReviews]
- [2] Zollino M, et al. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. 2003;72(3):590-597. [PubMed]
- [3] Stec I, et al. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum Mol Genet. 1998;7(7):1071-1082. [PubMed]
- [4] Endele S, et al. LETM1, a novel gene encoding a putative EF-hand Ca(2+)-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS) critical region and is deleted in patients with WHS. Genomics. 1999;60(2):218-225. [PubMed]
- [5] Battaglia A, et al. Natural history of Wolf-Hirschhorn syndrome: experience with 15 cases. Pediatrics. 1999;103(4 Pt 1):830-836. [PubMed]
- [6] OMIM #194190 – Wolf-Hirschhorn Syndrome. [OMIM]
- [7] Orphanet – Wolf-Hirschhorn Syndrome. [Orphanet]
- [8] GARD – Wolf-Hirschhorn Syndrome. National Institutes of Health. [GARD]
- [9] 難病情報センター – 4p欠失症候群(指定難病198). [難病情報センター]
- [10] MedlinePlus – Wolf-Hirschhorn Syndrome. [MedlinePlus]
関連記事







