目次

脊髄性筋萎縮症 下肢優位型 2A(SMALED2A)は、BICD2遺伝子のヘテロ接合性ミスセンス変異によって引き起こされる、常染色体顕性(優性)遺伝形式をとる希少な神経筋疾患です。乳幼児期から下肢優位の筋力低下や足部変形を呈する一方、進行は極めて緩徐で、多くの患者さんが生涯にわたって歩行能力を保ち、生命予後も健常者と変わりません。感覚や知的機能は障害されず、適切な多職種連携による管理によって生活の質を維持できることが、本疾患の最も重要な臨床的特徴です。
Q. SMALED2Aとはどのような病気ですか?まず結論だけ知りたいです
A. BICD2遺伝子の変異によって起こる、下肢の筋肉が優位に弱くなる遺伝性神経筋疾患です。乳幼児期に発症して大腿四頭筋などの太ももの筋肉が痩せていきますが、進行は非常にゆっくりで、知的機能や感覚は保たれ、寿命にも影響しません。SMN1遺伝子の変異による「5q-SMA」とは別の疾患で、治療薬の選択肢も異なります。
- ➤疾患の定義 → OMIM 615290、非5q-SMAに分類される常染色体顕性(優性)疾患
- ➤分子メカニズム → ダイニン複合体の「機能獲得型」異常活性化が病態の中核
- ➤主な症状 → 下肢優位の筋力低下・動揺性歩行・足部変形・関節拘縮
- ➤鑑別診断 → SMALED1・5q-SMA Type 4・遺伝性痙性対麻痺との違い
- ➤診断・管理 → BICD2遺伝子検査と多職種連携によるリハ・整形外科管理
1. SMALED2Aとは:疾患の定義と歴史的背景
SMALED2Aは「Spinal Muscular Atrophy, Lower Extremity-predominant, 2A(脊髄性筋萎縮症 下肢優位型 2A)」の略称です。OMIM 615290として国際的に登録されており、脊髄性筋萎縮症(SMA)の中でも、SMN1遺伝子の変異を原因としない「非5q-SMA」と呼ばれるグループに分類されます。
💡 用語解説:常染色体顕性(優性)遺伝
「常染色体」とは性染色体(X染色体・Y染色体)以外の染色体のことです。「顕性(けんせい)」は従来「優性(ゆうせい)」と呼ばれていた用語で、2本ある染色体のうち片方に変異があるだけで症状が現れる遺伝形式を意味します。SMALED2Aの場合、罹患した親から子に変異遺伝子が受け継がれる確率は理論上50%です。ただし、家族歴のない孤発例として新生突然変異(de novo変異)で生じるケースも報告されています。
5q-SMAの大部分が常染色体劣性(潜性)遺伝で、乳児期に重篤な運動機能障害をきたすのに対し、SMALED2Aは常染色体顕性(優性)遺伝形式をとり、病状は極めて緩徐に進行し、生命予後は健常者と同等です。多くの方が高齢期まで歩行能力を維持できるという点が、この疾患を理解するうえで最も重要なポイントです。
SMALED1とSMALED2の分離:歴史的経緯
下肢優位の常染色体顕性SMA(SMA-LED)の遺伝的原因が探索される過程で、初期の連鎖解析によって第14染色体長腕(14q32)に強い連鎖が示されました。この領域の原因遺伝子はその後DYNC1H1と同定され、「SMALED1(OMIM 158600)」と命名されました。
続いて2013年、第9染色体長腕(9q22.31)にマッピングされるBICD2遺伝子のヘテロ接合性変異を原因とする一群が、SMALED1とは別の疾患として独立した「SMALED2」として確立されました。さらにその後、より重篤な胎児期発症の表現型が「SMALED2B(OMIM 618291)」として分離され、本ページで扱う緩徐進行性のタイプが「SMALED2A」と呼ばれるようになっています。SMALED2Bについては別ページで詳しく解説しています。
🔍 関連記事
2. 原因遺伝子BICD2と分子病態メカニズム
BICD2遺伝子は、細胞内の物質輸送ネットワークにおいて「カーゴアダプター」と呼ばれる重要な役割を果たすタンパク質をコードしています。SMALED2Aの病態を理解するうえで欠かせない、ダイニン・ダイナクチン複合体との関係から見ていきましょう。
💡 用語解説:ダイニン・ダイナクチン複合体
細胞内には「微小管(びしょうかん)」というレールが張り巡らされており、その上を物質を運ぶモータータンパク質が走っています。ダイニンは細胞の末端から中心(核の方向)に向かって物資を運ぶ「逆行性輸送」を担うモーターで、ダイナクチンはそれを補助する複合体です。この2つは協調して働いており、特に長い軸索を持つ神経細胞では、この輸送が止まると細胞自体が機能しなくなります。
💡 用語解説:ミスセンス変異
DNAの塩基が1つ変化することで、タンパク質を構成するアミノ酸が別の種類に置き換わるタイプの変異です。「タンパク質の設計図」の1文字が変わるイメージで、タンパク質全体の形や機能に影響を与えることがあります。SMALED2Aの原因となるBICD2変異の多くはこのミスセンス変異です。
「機能獲得型」という独特な病態
SMALED2Aの分子病態を理解するうえで最も重要なポイントは、BICD2のミスセンス変異がタンパク質の機能を「失わせる」のではなく、むしろ「強めすぎる」方向に作用することです。これを「機能獲得(gain-of-function)」と呼びます。多くの遺伝性疾患は機能喪失(loss-of-function)が原因であり、機能獲得型の病態は治療戦略を考えるうえで全く異なるアプローチを必要とします。
💡 用語解説:機能獲得(gain-of-function)
変異したタンパク質が正常な機能を失うのではなく、本来の機能を過剰に発揮してしまうあるいは新しい異常な機能を獲得してしまう状態です。SMALED2Aの場合、変異型BICD2はダイニンとの結合親和性が異常に高まり、逆行性輸送が過剰に活性化されます。順行性輸送と逆行性輸送のバランスが崩れることで、神経突起の伸長が阻害され、運動ニューロンが徐々に変性していきます。
変異型BICD2を発現させた神経細胞では、ゴルジ体の断片化や神経突起の伸長阻害が観察されます。さらに、Bicd2遺伝子をノックアウトしたマウスの研究では、骨格筋組織におけるBICD2の喪失が運動ニューロンの変性を駆動する「細胞非自律的」なメカニズムも明らかになりつつあり、なぜ筋肉量の多い下肢に症状が集中するのかという臨床的疑問への分子レベルでの解答が見えてきました。
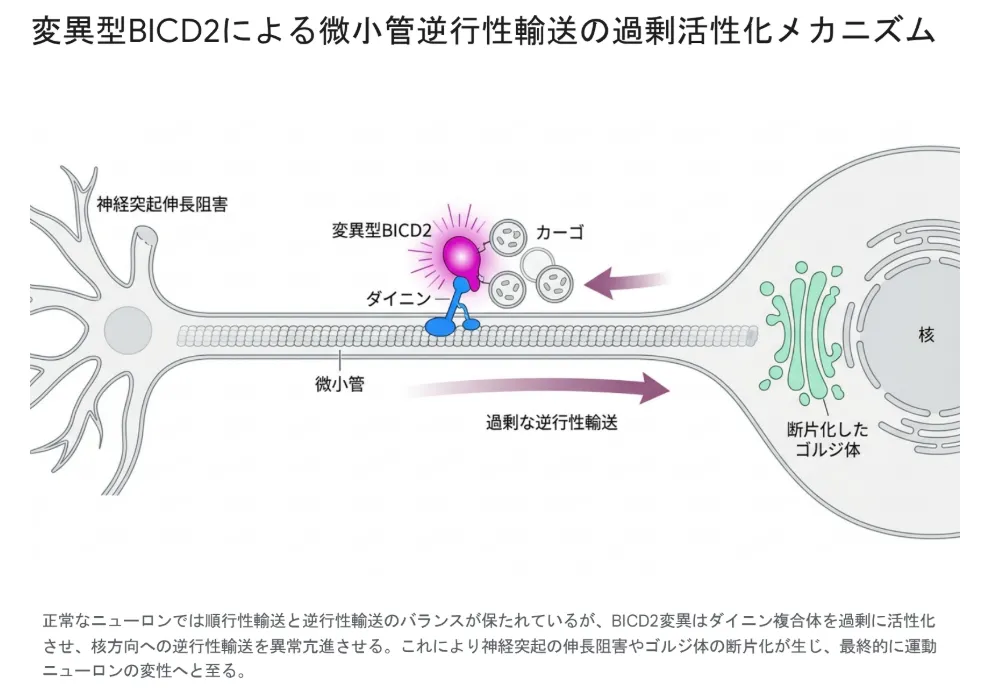
正常なニューロンでは順行性輸送と逆行性輸送のバランスが保たれているが、BICD2変異はダイニン複合体を過剰に活性化させ、核方向への逆行性輸送を異常亢進させる。これにより神経突起の伸長阻害やゴルジ体の断片化が生じ、最終的に運動ニューロンの変性へと至る。
3. 主な症状と表現型スペクトラム
SMALED2Aの臨床像は、その名の通り「下肢優位(lower extremity-predominant)」が最大の特徴です。症状の現れる時期や程度には個人差がありますが、典型的には乳幼児期から小児期早期にかけて顕在化します。
🦵 下肢の筋力低下
- 大腿四頭筋の著明な萎縮
- 股関節屈曲・膝関節伸展の困難
- 立ち上がり・階段昇降の困難
- Gowers徴候陽性(小児期)
🚶 歩行・姿勢の異常
- 動揺性歩行(waddling gait)
- つま先歩き(toe-walking)
- 歩行獲得の遅延
- 走行・階段昇降が困難
🦶 骨関節系の変形
- 内反尖足(クラブフット)
- 凹足(pes cavus)
- アキレス腱・膝関節の拘縮
- 股関節の異形成・脱臼の合併
✅ 保たれる機能(重要)
- 感覚機能
- 認知・知的機能
- 嚥下・発語(球機能)
- 呼吸機能(通常は影響軽微)
SMALED2Aの臨床像は「純粋な」運動ニューロン疾患の特徴を色濃く残しています。感覚障害・知的障害・嚥下障害・重篤な呼吸不全をきたさないことは、患者さんとご家族にとって極めて重要な予後情報です。深部腱反射は遠位で減弱・消失することが多いものの、一部の患者さんでは痙縮や腱反射の亢進といった上位運動ニューロン徴候を呈することも報告されており、表現型のばらつきが存在します。
BICD2関連疾患の重症度スペクトラム
BICD2遺伝子の変異が引き起こす疾患は、軽症のSMALED2Aから胎児期発症の重症型SMALED2B、さらには脳形成異常(滑脳症)まで、連続的なスペクトラムを形成することが近年明らかになっています。
BICD2関連疾患の重症度スペクトラム
← 軽症(緩徐進行性) ・・・・・・・・・・ 重症(胎児期発症・致死的) →
この3群は同じBICD2遺伝子の変異であっても、変異の部位や種類によって全く異なる臨床像を呈します。SMALED2Aを診断する際には、家系内に重症例や流産・死産歴がないかを丁寧に確認することが、本人の予後説明と家族計画の支援において重要です。

4. 鑑別診断:SMALED1・5q-SMA・遺伝性痙性対麻痺との違い
SMALED2Aは、下肢優位の筋萎縮や歩行障害といった非特異的な症状を呈するため、複数の神経筋疾患との鑑別が必要です。特に5q-SMA Type 4との誤診は歴史的に多く報告されており、正確な分子診断が予後説明と治療方針を左右します。
SMALED2Aと5q-SMA Type 4を臨床的に区別する最大のポイントは「初期の足部変形と拘縮の有無」です。SMALED2Aでは歩行開始時期の遅れとともに内反尖足などがすでに存在することが多いのに対し、5q-SMA Type 4ではこのような先天的な整形外科的異常は見られません。
発症時期と重症度の比較
下肢優位SMA関連疾患:発症時期×重症度の比較
赤色=重症型/青色=緩徐進行性/橙色=中等度/緑色=軽症型
🔍 関連記事
5. 診断アプローチと遺伝子検査
SMALED2Aの診断は、丁寧な臨床病歴の聴取と神経学的評価を出発点に、画像診断・電気生理学的検査・組織学的検査を経て、最終的に分子遺伝学的な確定診断に至る多角的なアプローチを必要とします。
臨床的レッドフラッグ:SMALED2Aを疑うサイン
💡 SMALED2Aを疑うべき臨床所見の組み合わせ
- ➤独歩獲得の遅れと下肢優位の筋力低下(特に大腿四頭筋)
- ➤動揺性歩行・つま先歩き・Gowers徴候陽性
- ➤内反尖足・凹足などの早期の足部変形
- ➤感覚・認知機能の保持(純粋運動ニューロン疾患の特徴)
- ➤常染色体顕性遺伝を示唆する家族歴の有無
電気生理学的検査・画像診断・生化学検査
針筋電図(EMG)
巨大運動単位電位の存在、慢性脱神経と再神経支配の証拠が確認され、神経原性変化を示します。下位運動ニューロン疾患の支持所見です。
筋生検
大腿四頭筋などからの生検で、群集萎縮・タイプ1線維優位性に加え、筋線維の縦裂や脂肪線維腫症様変化など筋原性所見も混在します。
下肢MRI
大腿部を中心に重度の脂肪変性が認められ、罹患筋の分布と重症度を視覚化できます。診断補助と進行評価の両面で有用です。
血液検査(CK)
血清クレアチンキナーゼ(CK)は正常範囲内、または中等度の軽微な上昇にとどまります。進行性筋ジストロフィーとの鑑別に有用です。
確定診断:BICD2遺伝子検査
💡 用語解説:新生突然変異(de novo変異)
両親には変異がなく、患者さん本人で新たに生じた遺伝子変異のことです。両親の生殖細胞(精子・卵子)または受精直後のごく早い段階で変異が起こります。SMALED2Aでは家族歴のない孤発例として新生突然変異が報告されており、ご両親に変異がなくても、お子さんに病気が現れるケースがあるということです。
最終的な確定診断は、BICD2遺伝子の病的バリアントの同定によって行われます。疾患の希少性と非定型的な症状を踏まえ、当院では脊髄性筋萎縮症(SMA)NGS遺伝子パネル検査(BICD2を含む29遺伝子を一度に解析)や、上位運動ニューロン徴候を伴う場合は遺伝性痙性対麻痺(HSP)NGS遺伝子パネル検査(BICD2/SPG51を含む38遺伝子)など、ターゲット遺伝子パネル検査を活用します。
遺伝子診断によってBICD2の病的バリアントが同定されることは、患者さんへの予後説明や、家族計画における遺伝カウンセリングを提供するうえで決定的な意味を持ちます。また、不完全浸透による無症候性キャリアの特定にも不可欠です。
6. 治療と長期管理プロトコル
現在のところ、SMALED2Aに対するFDA等で承認された疾患修飾治療薬は存在しません。5q-SMAに対するヌシネルセン(スピンラザ)、リスジプラム(エブリスディ)、オナセムノゲンアベパルボベク(ゾルゲンスマ)といった画期的な治療薬は、すべてSMN1遺伝子経路を標的としているため、BICD2変異によるSMALED2Aには効果がありません。
したがって、患者さんの長期的ケアにおいては、心臓・呼吸器・整形外科・リハビリテーションの専門家が集結する「多職種チーム(Multidisciplinary Team: MDT)」による包括的な症状管理と機能維持が、現時点での最大の治療戦略となります。
SMALED2Aの集学的ケアと介入戦略
🏃 理学療法・リハビリテーション
- 日々の関節可動域訓練(ストレッチ)
- 夜間スプリント(night splints)の活用
- スタンディングフレームの早期導入
- 労作後症状悪化(PESE)を考慮したペーシング
🦽 運動補助・装具療法
- 歩行器(walkers)による移動支援
- 側方支持を備えた専用車椅子
- 短下肢装具(AFO)の調整
- 学校環境のバリア解消支援
🦴 整形外科的サーベイランス
- 生後6〜18ヶ月から開始する年次股関節X線検査
- Reimer移動率(股関節変位)の定期評価
- 寛骨臼指数(Acetabular Index)の確認
- 頸体角・脊柱側弯の経過観察
🏥 外科的介入
- 大腿骨内反回転骨切り術(VDRO)
- 骨盤骨切り術(Pelvic osteotomy)
- アキレス腱延長術などの腱解離術
- 重度側弯症に対する脊柱変形矯正手術
💡 用語解説:労作後症状悪化(PESE)
Post-Exertional Symptom Exacerbationの略で、身体的または認知的な活動の後、12〜72時間遅れて筋力低下などの症状が急激に悪化し、回復に数日から数週間を要する現象です。SMALED2Aの臨床的特徴として報告されており、理学療法やリハビリテーションの強度設定では「やりすぎず、休みすぎない」適切なペーシングが極めて重要です。
予後と生活の質
SMALED2Aの臨床的予後は、一般に考えられているよりも遥かに良好です。本疾患における筋肉症状や関連する健康上の問題は、時間とともに進行(悪化)しないか、あるいは極めてゆっくりとしか進行しません。重症の5q-SMA Type 1やType 2とは異なり、呼吸不全などの致死的合併症をきたさず、生命予後は健常者と同等です。多くの方が50代・80代といった高齢期に至るまで歩行可能であることが文献で報告されています。
7. 遺伝カウンセリングの意義
SMALED2Aの確定診断後、ご家族への丁寧な遺伝カウンセリングが極めて重要になります。当院では臨床遺伝専門医が、医学的事実と心理的支援の両面からご家族に伴走します。
- ➤遺伝形式と再発リスクの説明:常染色体顕性(優性)遺伝のため、患者本人が子どもを持つ場合の遺伝確率は理論上50%です。一方、新生突然変異による孤発例も多く、両親に変異がない場合は次子の再発リスクは低いと考えられますが、生殖細胞モザイクの可能性も完全には除外できません。
- ➤不完全浸透への配慮:BICD2変異には不完全浸透(変異を持っていても症状が現れないケース)が報告されており、家族内で症状の有無や程度に差があることを説明します。
- ➤予後情報の提供:多くの方が生涯歩行可能で寿命にも影響しないという情報は、ご本人とご家族が今後の人生設計を立てるうえで非常に重要な希望の根拠となります。
- ➤出生前診断の選択肢:次子を希望される場合、既知の変異が同定されていれば絨毛検査・羊水検査による出生前遺伝子診断が選択肢として存在します。ただしSMALED2Aは緩徐進行性で生命予後良好な疾患であり、出生前に診断することが必ずしも利益になるとは限りません。検査を受けるかどうかはご家族の価値観と将来設計に基づいて十分にお話しする必要があります。
8. よくある誤解
誤解①「SMA=寿命が短い」
SMAという言葉から重症の5q-SMA Type 1(乳児期発症型)を連想する方が多いのですが、SMALED2Aは生命予後が健常者と変わらない緩徐進行性の疾患です。同じ「SMA」という名前でも、遺伝子も予後も全く異なります。
誤解②「スピンラザやゾルゲンスマで治せる」
5q-SMA向けに承認された疾患修飾薬は、すべてSMN1遺伝子経路を標的としています。BICD2変異によるSMALED2Aには効果がありません。遺伝子診断によって正しい疾患名を確定することが、不適切な治療を避けるうえで重要です。
誤解③「親に同じ症状がないから遺伝ではない」
SMALED2Aは新生突然変異(de novo変異)として孤発する例が多く、両親が健常でもお子さんに発症することがあります。また不完全浸透により、変異を持っていても症状が軽微で気づかれていないケースも存在します。
誤解④「SMALED1とSMALED2は同じ病気」
臨床像は酷似していますが、原因遺伝子が全く異なる(SMALED1はDYNC1H1、SMALED2はBICD2)別疾患です。遺伝子検査による鑑別が必須であり、家系内の遺伝学的評価にも影響します。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 SMALED2Aの遺伝子検査・遺伝カウンセリングについて
SMALED2Aをはじめとする希少遺伝性神経筋疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] OMIM #615290. Spinal Muscular Atrophy, Lower Extremity-Predominant, 2A, Autosomal Dominant. Johns Hopkins University. [OMIM 615290]
- [2] OMIM #618291. Spinal Muscular Atrophy, Lower Extremity-Predominant, 2B, Autosomal Dominant. [OMIM 618291]
- [3] Oates EC, et al. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet. 2013;92(6):965-973. [PMC2918478]
- [4] Martinez Carrera LA, Wirth B. Dominant spinal muscular atrophy is caused by mutations in BICD2, an important golgin protein. Front Neurosci. 2015;9:401. [PMC4633519]
- [5] Huynh W, et al. Disease-associated mutations in human BICD2 hyperactivate motility of dynein–dynactin. J Cell Biol. 2017;216(10):3051-3060. [J Cell Biol]
- [6] Storbeck M, et al. Phenotypic extremes of BICD2-opathies: from lethal, congenital muscular atrophy with arthrogryposis to asymptomatic with subclinical features. Eur J Hum Genet. 2017;25(9):1040-1048. [PMC5558181]
- [7] Koboldt DC, et al. The Genotypic and Phenotypic Spectrum of BICD2 Variants in Spinal Muscular Atrophy. Ann Neurol. 2020;87(4):487-496. [Wiley]
- [8] Rudnik-Schöneborn S, et al. Loss of BICD2 in muscle drives motor neuron loss in a developmental form of spinal muscular atrophy. Acta Neuropathol Commun. 2020;8(1):14. [PMC7076953]
- [9] Tsai MH, et al. Impairment in dynein-mediated nuclear translocation by BICD2 C-terminal truncation leads to neuronal migration defect and human brain malformation. Acta Neuropathol Commun. 2020;8(1):106. [PMC7362644]
- [10] MedlinePlus Genetics. Spinal muscular atrophy with lower extremity predominance. National Library of Medicine. [MedlinePlus]






