目次
8q22.1欠失症候群(ナブルスマスク様顔面症候群)とは?
症状・原因・診断・治療を臨床遺伝専門医が解説
Q. 8q22.1欠失症候群とはどのような病気ですか?
A. 8番染色体長腕(8q22.1)領域の微細な欠失によって生じる、極めて希少な先天性遺伝性症候群です。
ナブルスマスク様顔面症候群(NMLFS)とも呼ばれ、マスク様顔貌・眼瞼裂狭小・屈指症・停留精巣などの特徴的な症状が見られます。世界でわずか約26例しか報告されていない超希少疾患です。
-
➤
原因 → 8番染色体長腕8q21.3-q22.1領域の約2.78〜4Mbの微小欠失 -
➤
主要症状 → マスク様顔貌、眼瞼裂狭小、屈指症、停留精巣(男児でほぼ必発)、発達遅滞 -
➤
重要な特徴 → 「幸福そうな気質」:表情筋が動きにくいにもかかわらず明るく社交的 -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
頻度 → 100万人に1人未満(世界で約26例のみ報告)
1. 8q22.1欠失症候群とは|基本情報
【結論】 8q22.1欠失症候群(ナブルスマスク様顔面症候群:NMLFS)は、8番染色体長腕の8q21.3-q22.1領域が欠失することで発症する、極めて希少な先天性染色体異常症候群です。世界で約26例しか報告されておらず、有病率は100万人に1人未満と推定されています。
「マスク様顔貌」という独特の名称は、患者さんの顔面が能面(マスク)のように表情に乏しく見えることに由来しています。しかし、この特徴的な顔貌は全例に見られるわけではなく、近年の研究ではより広範な全身性疾患であることが明らかになっています。

8番染色体における8q22.1領域の位置
💡 用語解説:「微小欠失症候群」とは?
染色体の一部が欠損する異常で、通常の染色体検査(G分染法)では検出できないほど小さな欠失(数百kb〜数Mb)を指します。このような微小欠失は、染色体マイクロアレイ検査(CMA)などの高解像度検査でのみ検出可能です。欠失領域に含まれる複数の遺伝子の機能が失われることで、様々な症状が現れます。
8q22.1欠失症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 8q22.1微細欠失症候群(OMIM #608156) |
| 別名 | ナブルスマスク様顔面症候群(Nablus Mask-Like Facial Syndrome; NMLFS) |
| 原因 | 8q21.3-q22.1領域の約2.78〜4Mbの微小欠失 |
| 頻度 | 100万人に1人未満(世界で約26例) |
| 遺伝形式 | 常染色体優性(顕性)・多くは新生突然変異 |
| 主要候補遺伝子 | GDF6、ZFPM2、CCNE2、FAM92A1 |
発見の歴史
本症候群の歴史は21世紀初頭に始まります。2000年、パレスチナの遺伝学者Ahmad Teebi博士が、ナブルス出身の4歳男児において特異な顔貌と臨床徴候の組み合わせを報告しました。「ナブルス」という名称は、この最初の患者の出身地に由来しています。

8q22.1欠失症候群の発見から解明までの年表
📅 発見から解明への道のり
2000年:Teebi博士が最初の症例を報告し、NMLFSと命名
2003年:Salpietroらが2例目を報告、独立した症候群として確認
2006年:Shiehらがアレイ CGHで8q21.3-q22.1の共通欠失を同定
2009年:Raas-Rothschildらが責任領域を2.78Mbに絞り込み
2024年:35歳男性の症例報告で成人期の進行性ミオパチーが示唆される
⚠️ 重要な発見:欠失だけでは説明できない表現型の多様性
2018年までの研究で、8q22.1欠失が確認された26例のうち、典型的なマスク様顔貌を呈したのは半数の13例のみであることが判明しました。これは、欠失が発症の「必要条件」ではあるが「十分条件」ではない可能性を示唆しています。修飾遺伝子やエピジェネティック要因の関与が疑われています。
2. 8q22.1欠失症候群の主な症状
【結論】 本症候群の臨床像は、マスク様顔貌を中心とした頭蓋顔面の特徴、骨格系(屈指症・関節拘縮)、神経発達(発達遅滞・知的障害)、泌尿生殖器(停留精巣)の4つの主要な領域にわたります。また、「幸福そうな気質」という特徴的な行動特性も報告されています。
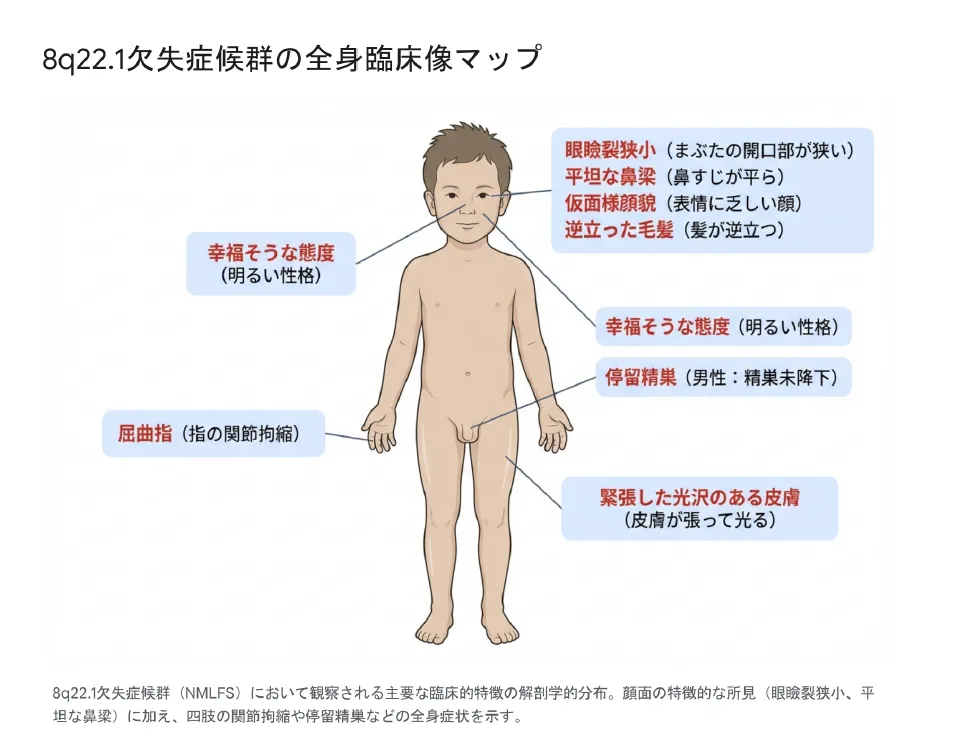
8q22.1欠失症候群の主要な臨床的特徴の解剖学的分布
頭蓋顔面の特徴(Craniofacial Phenotype)
本症候群における頭蓋顔面の異形成は、診断の最も強力な手がかりとなります。「マスク様」と形容される顔貌は、神経原性の顔面神経麻痺とは質的に異なる特徴を示します。
-
•
マスク様顔貌:静止時に無表情で、笑ったり泣いたりする際の表情筋の動きが著しく制限
-
•
皮膚の光沢:顔面の皮膚が薄く張りつめ、ガラスのような独特の光沢を放つ
-
•
眼瞼裂狭小:目の開口幅が狭く、目が小さく見える
-
•
平坦な鼻梁:鼻根部が平坦で幅広く、鼻尖が球状で肉厚
-
•
その他:眉毛・睫毛が疎、眼間開離、小顎症、低位耳介
💡 用語解説:「眼瞼裂狭小(Blepharophimosis)」とは?
眼瞼裂狭小は、上下のまぶたの開口部(眼瞼裂)の縦径と横径がともに短縮している状態です。単なるまぶたの皮膚の被さりではなく、眼瞼の構造的な発育不全を示唆します。本症候群では高頻度で認められる重要な所見です。
筋骨格系の異常
骨格および関節の異常は、顔貌に次いで高頻度に認められる所見であり、患者さんの身体機能に直接的な影響を及ぼします。
屈指症(Camptodactyly)
手指の近位指節間関節(PIP関節)が屈曲位で拘縮し、完全な伸展が不能となる状態。特に第5指(小指)に好発しますが、全指に及ぶこともあります。
多発性関節拘縮
手指に限らず、手首・肘・股関節・膝・足首など全身の関節に拘縮が見られることがあります。重症例では先天性多発性関節拘縮症様の外観を呈します。
神経発達と行動特性
神経発達への影響は必発ですが、その重症度には個人差があります。
| 神経発達の特徴 | 詳細 |
|---|---|
| 知的発達 | 軽度〜中等度の知的障害が多い。重度精神遅滞は比較的少ない |
| 運動発達 | 首のすわり、独歩の獲得が遅れる傾向。多くは最終的に歩行を獲得 |
| 言語発達 | 言語表出の開始が遅れるが、多くは会話を獲得 |
| 行動特性 | 「幸福そうな気質(Happy Demeanor)」:社交的で明るく愛想が良い |
| その他 | 小頭症、筋緊張低下(乳児期)、一部でADHD・学習障害の合併 |
🌟 特徴的な「幸福そうな気質」
本症候群の患者さんの多くは、顔面の表情筋が動きにくいにもかかわらず、概して社交的で明るい性格を持っています。「顔は笑っていないが、雰囲気や行動は楽しげである」という独特のパラドックスが特徴です。この特性はアンジェルマン症候群の「幸福な人形」様相と一部重複しますが、NMLFSではより穏やかで多動や興奮を伴わない場合が多いとされています。
泌尿生殖器・歯科的合併症
停留精巣(男児でほぼ必発)
- •
男児においてほぼ全例に認められる重要所見
- •
陰嚢低形成や小陰茎を伴うことも
- •
ZFPM2遺伝子の機能不全が関与
歯科的異常
- •
エナメル質形成不全:歯が脆弱で虫歯リスク高
- •
歯列不正、歯の欠損
- •
高口蓋に伴う咬合異常

🩺 院長コラム【2024年の新知見:成人期の進行性ミオパチー】
2024年に報告された35歳男性の症例は、本症候群の長期的な理解に重要な知見をもたらしました。この患者さんでは、青年期以降に進行性の筋力低下(ミオパチー)が出現し、21歳の筋生検でミオパチーに合致する病理学的変化が確認されました。
これは、8q22.1欠失症候群が単なる「先天的な形態異常」にとどまらず、加齢とともに進行する側面を持つ可能性を示唆しています。成人期の患者さんには、定期的な神経筋機能のモニタリングが必要かもしれません。まだ1例の報告ですので、今後のデータ蓄積が待たれます。
3. 原因と遺伝的背景|主要候補遺伝子
【結論】 本症候群の原因は、8q21.3-q22.1領域(約2.78Mb)に位置する複数の遺伝子のハプロ不全です。主要な候補遺伝子としてGDF6(骨格形成)とZFPM2(心臓・性腺発生)が注目されていますが、欠失があっても症状が出ない例も多く、遺伝子型と表現型の関係は複雑です。
💡 用語解説:「ハプロ不全(Haploinsufficiency)」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。ハプロ不全とは、染色体の欠失などにより1コピーが失われ、残り1コピーだけでは正常な機能を維持できない状態を指します。遺伝子産物(タンパク質)が通常の約50%に減少することで、細胞や臓器の発生・機能に異常が生じます。
主要な候補遺伝子とその機能
| 遺伝子 | 主な機能 | 関連する臨床症状 |
|---|---|---|
| GDF6 | 骨形成因子(BMPファミリー)、頭蓋縫合・関節形成の制御 | 眼瞼裂狭小、平坦な鼻梁、屈指症、関節拘縮 |
| ZFPM2 | GATA転写因子のコファクター、心臓・横隔膜・性腺の発生制御 | 停留精巣、鼠径ヘルニア、心血管異常 |
| CCNE2 | 細胞周期のG1/S移行制御(サイクリンE2) | 精子形成障害、性腺機能低下の可能性 |
| FAM92A1 | 繊毛形成、ミトコンドリア機能(BARドメインタンパク) | 特徴的顔貌への関与が示唆 |
GDF6:骨格形成における中心的役割
-
①
骨形成因子(BMP)ファミリー:TGF-βシグナル伝達経路を通じて骨・軟骨・関節の形成を制御
-
②
頭蓋縫合の制御:マウスモデルでGdf6欠損は頭蓋骨の癒合不全を引き起こす
-
③
他の疾患との関連:クリッペル・ファイル症候群など他の骨格系疾患にも関与
遺伝子型と表現型の不一致
本症候群における最大の謎の一つは、同じ8q22.1欠失を持ちながら症状が大きく異なることです。
🎯 「必要条件だが十分条件ではない」仮説
Allansonら(2012年)は、8q22.1欠失は本症候群発症の「必要条件」ではあるが「十分条件」ではないと提唱しました。
典型的なNMLFS表現型が完成するためには、以下の付加的要因が必要と考えられています:
• 修飾遺伝子の変異(欠失領域外の遺伝子多型など)
• エピジェネティックな要因(メチル化パターンの変化など)
• 環境要因や他の遺伝的背景との相互作用
この仮説は、同じ欠失を持つ無症状の親から重度の症状を持つ子が生まれるといった家族内変動を説明する上でも重要です。
欠失の発生機序と遺伝形式
新生突然変異(de novo)
多くの症例は新生突然変異として発生します。両親は正常で、配偶子形成時または受精後初期に新たに欠失が生じます。家族内再発はまれです。
家族性継承
一部では、無症状の親から欠失を受け継ぐケースが報告されています。遺伝形式は常染色体優性(顕性)ですが、不完全浸透のため親が無症状のことがあります。
4. 8q22.1欠失症候群の診断方法
【結論】 本症候群の確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のG分染法(核型分析)では、数Mb程度の微細欠失は検出限界以下となることが多く、見逃されるリスクが高いです。臨床症状のみで診断することはできません。
診断の契機となる臨床所見
-
①
マスク様顔貌+光沢のある皮膚(特異性が高い)
-
②
眼瞼裂狭小+平坦な鼻梁+屈指症
-
③
男児での停留精巣+上記の顔面異常
-
④
発達遅滞+「幸福そうな気質」
遺伝学的検査の種類
| 検査方法 | 特徴 | 8q22.1欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。数kb〜数Mbの微細CNVを高解像度で検出 | ◎ 検出可能 |
| G分染法(核型分析) | 解像度は5〜10Mb程度。大きな転座や数的異常を検出 | ✕ 検出困難(約2〜4Mbの微小欠失) |
| FISH法 | 特定領域のプローブを使用。家族検査に有用 | △ 専用プローブで可能 |
| 全エクソーム解析(WES) | 非典型例の鑑別に有用。欠失が偶発的に発見されることも | △ CNV解析併用で可能 |
💡 用語解説:染色体マイクロアレイ(CMA)とは?
CMAは、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。日本では原因不明の発達遅滞・先天異常に対する保険適用検査として実施されています。アレイCGH(比較ゲノムハイブリダイゼーション)やSNPアレイなどの技術が用いられます。
鑑別診断
8q22.1欠失症候群は、いくつかの類似疾患と臨床像が重複するため、慎重な鑑別が必要です。
| 鑑別疾患 | 共通点 | 相違点 |
|---|---|---|
| 眼瞼-鼻-顔面症候群(BNFS) | 眼瞼裂狭小、マスク様顔貌、関節異常 | BNFSでは関節過伸展(NMLFSでは拘縮)、涙管閉塞が特徴的 |
| 8q21.11微細欠失症候群 | 同じ8番染色体長腕の欠失 | 丸い顔、眼瞼下垂、人中短縮(NMLFSでは平坦で長い人中) |
| メビウス症候群 | 先天性無表情 | 神経原性(顔面神経麻痺)。外転神経麻痺を伴う |
| フリーマン・シェルドン症候群 | 手指の屈曲変形 | 「口笛を吹くような顔」。原因遺伝子はMYH3 |
5. 治療と長期管理
【結論】 本症候群には根本的な治療法は存在せず、症状に応じた対症療法・早期療育・継続的支援が中心となります。疾患の多臓器性に鑑み、小児科医をコーディネーターとした集学的チーム医療が不可欠です。
ライフステージに応じた管理
| ライフステージ | 主な対応 |
|---|---|
| 乳児期・幼児期(0〜5歳) | 停留精巣の手術(精巣固定術)、口蓋裂の手術、早期療育開始(PT・OT・ST)、摂食嚥下指導 |
| 学童期・思春期(6〜18歳) | 個別の教育支援計画(IEP)、特別支援教育、行動・心理的支援、歯科的管理(エナメル質形成不全対策) |
| 成人期(19歳〜) | 神経筋モニタリング(ミオパチーのリスク)、就労・生活支援、心機能モニタリング |
症状別の治療・対応
停留精巣
- •
生後6ヶ月〜1歳頃に精巣固定術
- •
妊孕性温存・精巣腫瘍リスク低減が目的
- •
小児泌尿器科との連携
屈指症・関節拘縮
- •
ストレッチ・装具療法
- •
作業療法(OT)による巧緻性訓練
- •
必要に応じて腱延長術・関節授動術
発達遅滞・知的障害
- •
早期療育が最も重要
- •
理学療法(PT)・言語聴覚療法(ST)
- •
特別支援教育の活用
歯科的管理
- •
高濃度フッ素塗布・シーラント
- •
う蝕予防の徹底
- •
矯正歯科との連携
⚠️ 成人期の重要な注意点:2024年の35歳男性症例報告により、成人期に進行性のミオパチー(筋疾患)が出現する可能性が示唆されました。成人患者さんでは、原因不明の筋力低下や易疲労性がある場合、神経内科的な精査を検討してください。
6. 遺伝カウンセリングの重要性
【結論】 8q22.1欠失症候群の不完全浸透と表現型の多様性は、遺伝カウンセリングを複雑にします。「欠失=必ず発症」ではないこと、予後予測が困難であることを丁寧に説明し、家族の意思決定を支援することが重要です。
遺伝カウンセリングで伝えるべきポイント
-
①
表現型の多様性:同じ欠失でも、典型的なマスク様顔貌を呈するのは約半数のみ
-
②
予後の不確実性:発症の有無・重症度を出生前に予測することは困難
-
③
両親の検査:親が無症状の保因者か確認することで情報提供に役立つ
-
④
長期フォローの必要性:成人期のミオパチーリスクも含めた継続的支援
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常(新生突然変異) | 1%未満(生殖細胞モザイクの可能性を考慮) |
| 片親が保因者 | 50%(ただし症状発現は不確実) |
🩺 院長コラム【遺伝カウンセリングで大切にしていること】
8q22.1欠失症候群の遺伝カウンセリングで最も難しいのは、「予後が予測できない」という不確実性をどうお伝えするかです。世界で約26例しか報告がなく、しかもそのうち半数は典型的な症状を示さないという現実があります。
「欠失があるから病気」でも「欠失があっても大丈夫」でもなく、「現時点ではわからないことが多い」という事実を正直にお伝えしています。特に出生前診断で見つかった場合、ご家族は大きな不安を抱えます。
臨床遺伝専門医として、中立的な立場で正確な情報を提供し、どのような決断をされてもサポートを続けることをお約束しています。不安を抱えている方は、ぜひ一度ご相談ください。
7. 出生前診断について|NIPTと羊水検査
【結論】 8q22.1欠失は出生前診断で検出可能ですが、NIPTでの検出には限界があります。羊水検査・絨毛検査での染色体マイクロアレイ検査が確定診断のゴールドスタンダードです。ただし、胎児の予後予測は困難であり、出生前診断で見つかった場合の対応には慎重な遺伝カウンセリングが必要です。
出生前検査での検出方法
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| NIPT(微小欠失検査を含む) | △ 限定的 | 約2〜4Mbの欠失は検出困難なことが多い。スクリーニング検査 |
| 羊水検査+CMA | ◎ 検出可能 | 確定診断のゴールドスタンダード |
| 絨毛検査+CMA | ◎ 検出可能 | 妊娠初期(11〜14週)に実施可能 |
⚠️ ミネルバクリニックの微小欠失検査について
当院のCOATE法を用いたNIPTでは12種類の微小欠失を検査対象としていますが、8q22.1欠失は検査対象外です(対象は8q23q24 delであり、8q22.1とは異なる領域です)。8q22.1欠失の確定診断には羊水検査・絨毛検査での染色体マイクロアレイ検査が必要です。
出生前診断で見つかった場合の対応
-
①
遺伝カウンセリング:欠失の意味、表現型の多様性、予後の不確実性を説明
-
②
両親の検査:親が同じ欠失を持つか確認(保因者の有無は参考情報に)
-
③
詳細超音波:顔面異常、心奇形、骨格異常などを精査
-
④
出生後フォロー体制:発達モニタリング、早期療育の準備
⚠️ 重要な考慮点:出生前診断で8q22.1欠失が見つかった場合、「典型的な症状が出るかどうかは予測できない」という現実があります。超音波検査で明らかな異常がなくても症状が出る可能性がありますし、逆に欠失があっても軽症または無症状のこともあります。この不確実性をどう受け止めるかは、ご家族によって異なります。
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。全染色体検査や微小欠失検査も対応可能です。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会で費用面も安心
互助会制度(8,000円)により、陽性時の確定検査(羊水検査)費用を全額カバー。上限なしで安心です。
一人で悩まず、専門医を頼ってください
8q22.1欠失症候群について詳しく知りたい方、
出生前検査を検討している方は臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
🏥 一人で悩まないでください
8q22.1欠失症候群について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
参考文献
- [1] Teebi AS. Nablus mask-like facial syndrome. Am J Med Genet. 2000;95(4):407-408. [PubMed]
- [2] Shieh JT, et al. Nablus mask-like facial syndrome is caused by a microdeletion of 8q detected by array-based comparative genomic hybridization. Am J Med Genet A. 2006;140(12):1267-1273. [PubMed]
- [3] Raas-Rothschild A, et al. The 8q22.1 microdeletion syndrome or Nablus mask-like facial syndrome: report on two patients and review of the literature. Eur J Med Genet. 2009;52(2-3):140-144. [PubMed]
- [4] Allanson JE, et al. Nablus mask-like facial syndrome: deletion of chromosome 8q22.1 is necessary but not sufficient to cause the phenotype. Am J Med Genet A. 2012;158A(9):2091-2099. [PubMed]
- [5] Debost-Legrand A, et al. A new case of 8q22.1 microdeletion restricts the critical region for Nablus mask-like facial syndrome. Am J Med Genet A. 2013;161A(1):162-165. [PubMed]
- [6] Allanson JE, et al. Nablus syndrome: Easy to diagnose yet difficult to solve. Am J Med Genet C Semin Med Genet. 2018;178(4):447-457. [PubMed]
- [7] Mitrakos A, et al. Nablus mask-like facial syndrome: Report of an atypical case with 8q21.3-q22.1 deletion. Am J Med Genet A. 2024;194(10):e63826. [PubMed]
- [8] Yilmaz S, et al. Intraoral findings of a patient with Nablus mask-like facial syndrome and dental treatment approaches: a case report and literature review. J Clin Pediatr Dent. 2023;47(3):73-79. [PubMed]
- [9] OMIM #608156 – Nablus Mask-Like Facial Syndrome. [OMIM]
- [10] Orphanet: 8q22.1 microdeletion syndrome (ORPHA:178303). [Orphanet]
関連記事







