目次

Q. 17q12微小欠失症候群とはどのような病気ですか?
A. 第17染色体長腕(17q12)の約1.4Mb領域が欠失することで、腎臓・尿路異常や若年発症型糖尿病(MODY5)、神経発達障害などを引き起こす希少疾患です。
主要原因遺伝子であるHNF1Bの欠失により、腎嚢胞・高血糖・発達遅滞などの多彩な症状が生じますが、症状の出方には大きな個人差(多様性)があります。
-
➤
原因 → 17番染色体q12領域の約1.4Mb微小欠失(HNF1B, LHX1を含む) -
➤
主要症状 → 腎尿路奇形(90%)、神経発達障害(50%)、MODY5(40%)、MRKH症候群 -
➤
重要な特徴 → 可変的表現度:同じ欠失を持つ家族内でも、症状の種類や重症度が大きく異なる -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断の第一選択 -
➤
遺伝 → 常染色体顕性遺伝(優性遺伝)だが、約75%は新生突然変異(de novo)で発生
1. 17q12微小欠失症候群とは|基本情報と頻度
【結論】 17q12微小欠失症候群は、腎嚢胞などの腎異常、若年発症型糖尿病(MODY5)、神経発達症を三主徴とする染色体異常です。一般集団での頻度は約1/14,000〜1/50,000と希少ですが、腎奇形を持つ胎児や若年糖尿病患者の中ではより高頻度に見つかります。

この症候群は、かつては「腎嚢胞・糖尿病症候群(RCAD)」やHNF1B関連疾患として知られていましたが、近年では神経発達への影響や女性生殖器奇形などを含む全身性疾患として再定義されています。一つの臓器だけでなく、体全体の管理が必要となる病気です。
疾患の概要データ
| 項目 | 内容 |
|---|---|
| 疾患名 | 17q12微小欠失症候群(17q12 microdeletion syndrome) |
| OMIM番号 | #614527 |
| 欠失領域 | 17番染色体長腕 q12領域(約1.4Mb) |
| 推定頻度 | 約1/14,000 〜 1/50,000(集団により異なる) |
| 遺伝形式 | 常染色体顕性遺伝(優性遺伝) ※約75%は新生突然変異(de novo) |
| 主要原因遺伝子 | HNF1B、LHX1 |
2. 原因とメカニズム|HNF1BとLHX1遺伝子
【結論】 本症候群は、17q12領域にある約15個の遺伝子がまとめて失われることで発症します。特にHNF1B(腎・膵臓形成のマスター遺伝子)とLHX1(脳・生殖器形成に関与)の2つの遺伝子の機能不全(ハプロ不全)が、多様な症状の主原因です。
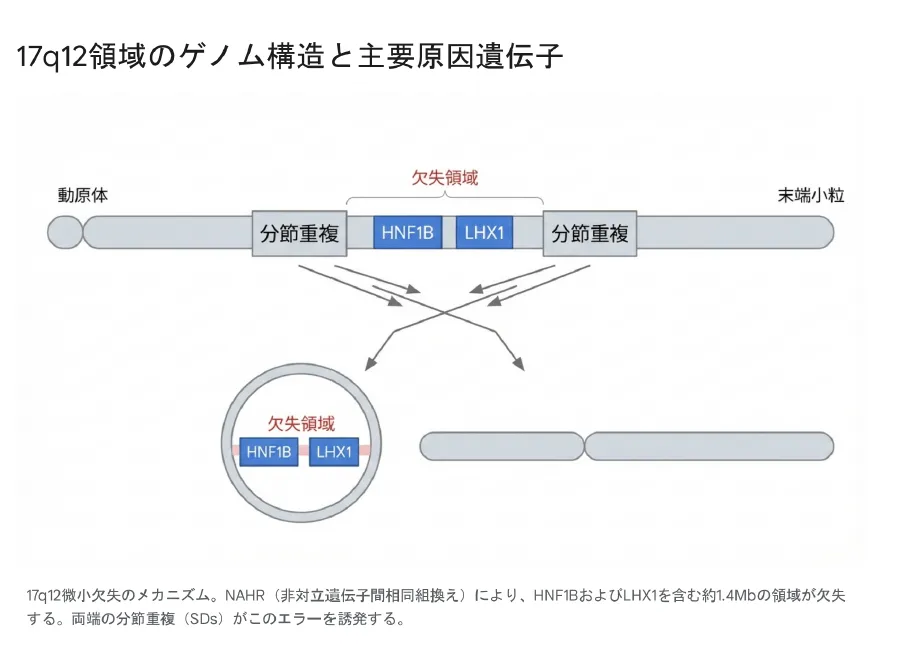
なぜ欠失が起きるのか?
17q12領域の両端には、DNA配列が非常によく似た「分節重複(LCRs)」という構造が存在します。卵子や精子が作られる際(減数分裂)、この似た配列同士が誤って結びつき、その間の領域が抜け落ちてしまうエラーが起こりやすくなっています。これを非対立遺伝子間相同組換え(NAHR)と呼びます。
主要な責任遺伝子の働き
HNF1B 遺伝子
- •
役割:腎臓、膵臓、肝臓、尿路の発生を指揮する司令塔
- •
欠失時の影響:
– 腎嚢胞、腎低形成
– MODY5型糖尿病
– 低マグネシウム血症
LHX1 遺伝子
- •
役割:脳神経系の発達、女性生殖器(ミュラー管)の形成
- •
欠失時の影響:
– 知的障害、ASD、統合失調症
– MRKH症候群(子宮・膣欠損)
💡 用語解説:ハプロ不全とは?
ヒトは通常、遺伝子を2セット(父母由来)持っています。1セットが欠失して残り1セットだけになったとき、作られるタンパク質の量が足りず、正常な機能を維持できなくなる状態を「ハプロ不全」といいます。17q12欠失症候群はまさにこのメカニズムで発症します。
3. 17q12微小欠失症候群の主な症状
【結論】 症状は「腎・尿路系」「代謝系」「神経系」「生殖器系」の4つに大別されます。最も頻度が高いのは腎異常ですが、成人期になって初めて糖尿病や精神疾患で診断されるケースもあります。
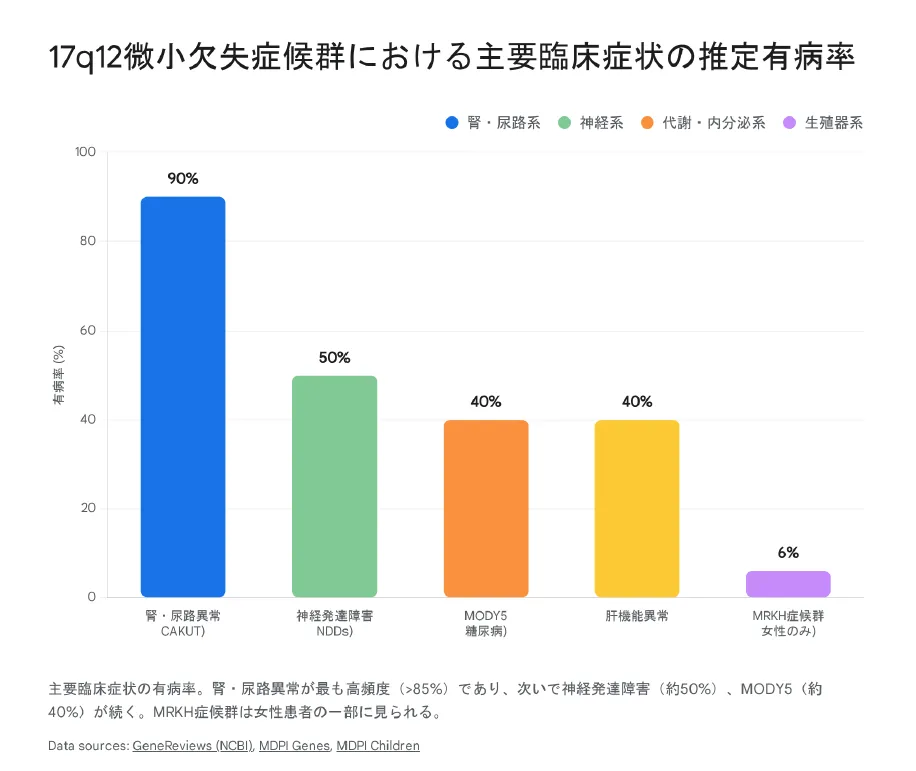
① 腎臓・尿路異常(CAKUT):約90%
患者さんの約9割に何らかの腎尿路異常が見られます。これが診断の最大のきっかけとなります。
- ● 腎嚢胞(Renal Cysts):最も特徴的。腎皮質に小さな嚢胞が多数できることが多く、多発性嚢胞腎とは異なります。
- ● 胎児期の高輝度腎:超音波検査で腎臓が白く輝いて見える所見(hyperechogenic kidneys)は、本症候群を疑う重要なサインです。
- ● その他:単腎(片方がない)、腎低形成(小さい)、馬蹄腎、水腎症など。
- ● 低マグネシウム血症:腎臓での再吸収障害により、血液中のマグネシウムが低下します。痙攣の原因になることもあります。
② 若年発症型糖尿病(MODY5):約40%
HNF1B欠失により膵臓の発育が悪くなり(膵低形成)、インスリン分泌が不足します。
- ● 発症時期:典型的には25歳未満(思春期〜若年成人期)に発症します。
- ● 特徴:肥満はなく、インスリン抵抗性もありません。1型や2型糖尿病と誤診されやすいですが、「腎嚢胞+若年糖尿病」があればMODY5を強く疑います。
③ 神経発達症・精神疾患:約50%
半数の患者さんで、発達や行動面での課題が見られます。LHX1遺伝子の影響が考えられています。
- ● 発達遅滞:言葉の遅れや学習障害(LD)。知的障害は軽度〜中等度が多いですが、正常範囲の方もいます。
- ● 自閉スペクトラム症(ASD):リスクが一般の数倍〜10倍高いとされています。
- ● 精神疾患:思春期以降、統合失調症や双極性障害、不安障害を発症するリスクが高まります。
④ 女性生殖器奇形(MRKH症候群):一部
女性患者の一部で、子宮や膣の上部が生まれつき欠損する「Mayer-Rokitansky-Küster-Hauser(MRKH)症候群」を合併します。15〜16歳になっても初経が来ない(原発性無月経)ことで発見されることが多いです。

🩺 院長コラム【症状の多様性について】
17q12微小欠失症候群の最大の特徴は、「人によって症状が全く違う」ということです。
重い腎不全で生まれるお子さんもいれば、大人になるまで無症状で気づかれない方もいます。また、腎臓は悪くても知能は全く正常な方もいれば、その逆もあります。
「欠失がある=必ず重症」ではありません。ご家族が同じ欠失を持っていても、症状の程度が親子で異なることもよくあります。この「不確実性」を理解した上で、定期的な検診で変化を早期に見つけることが管理のカギとなります。
4. 17q12微小欠失症候群の診断方法
【結論】 確定診断には染色体マイクロアレイ検査(CMA)が必須です。通常の染色体検査(Gバンド法)では微細な欠失を見逃す可能性が高いためです。胎児期、小児期、成人期それぞれのタイミングで診断の契機があります。
診断の契機となるサイン
👶 胎児期・乳児期
- ● 胎児エコーでの高輝度腎(腎臓が白い)
- ● 腎嚢胞、水腎症
- ● 羊水過多(多尿による)
🧒 小児期・学童期
- ● 原因不明の発達遅滞、言葉の遅れ
- ● 自閉スペクトラム症(ASD)
- ● 低マグネシウム血症
🧑 思春期・成人期
- ● 若年発症の糖尿病(MODY5)
- ● 原発性無月経(MRKH症候群)
- ● 統合失調症などの精神疾患
推奨される遺伝学的検査
| 検査方法 | 特徴 | 17q12欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。ゲノム全域の微細な欠失・重複を網羅的に解析可能。 | ◎ 確実に検出可能 |
| G分染法(核型分析) | 従来の染色体検査。5〜10Mb以上の大きな変化しか見えない。 | ✕ 検出困難(見逃されることが多い) |
| MLPA法 / FISH法 | HNF1B遺伝子など特定の場所を狙い撃ちで調べる。 | ○ ターゲット検査として有効 |
5. 治療とライフステージ別管理
【結論】 根本的な遺伝子治療法はありませんが、症状に応じた早期介入と管理により、QOL(生活の質)と予後は大きく改善します。腎臓、糖尿病、精神面の定期的なサーベイランスが生涯にわたり重要です。
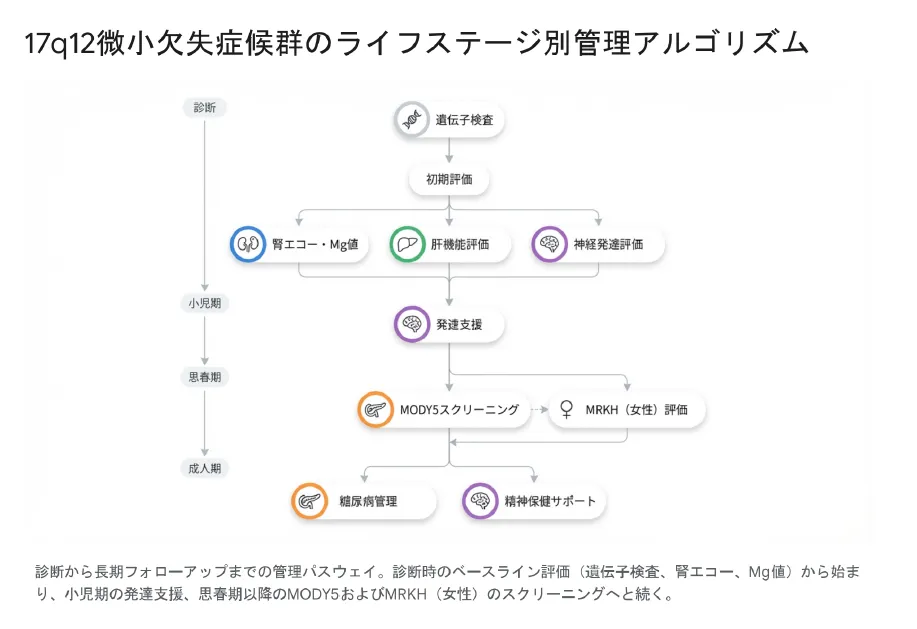
ライフステージ別のチェックポイント
| 時期 | 管理のポイント |
|---|---|
| 診断時 | ベースライン評価: 腎エコー、血液検査(クレアチニン、Mg、肝酵素)、尿検査、発達評価 |
| 小児期 | 腎機能・Mgモニタリング(年1回) 発達支援・療育(必要に応じて早期から) ASD/ADHDのスクリーニング |
| 思春期 | MODY5スクリーニング:HbA1c、血糖値(年1回) 女性:無月経の場合、産婦人科で子宮・膣の評価 精神症状(統合失調症の前駆症状など)の注意 |
| 成人期 | 腎不全の進行管理、糖尿病管理(インスリン等) 精神保健サポート、遺伝カウンセリング(次子計画) |
各症状への対応策
腎臓・代謝
- •
腎保護:血圧管理、腎毒性薬剤の回避
- •
低Mg血症:マグネシウム製剤の補充
- •
糖尿病:インスリン療法が標準的(スルホニル尿素薬が効くことも)
神経・精神
- •
発達支援:言語療法、作業療法、特別支援教育
- •
精神症状:薬物療法やカウンセリング
- •
注意:一部の精神科薬は代謝異常を起こしやすいため、糖尿病リスクのある本疾患では慎重に選択
6. 遺伝形式と次子への再発リスク
【結論】 17q12欠失は常染色体顕性遺伝(優性遺伝)ですが、約75%は両親が正常な「新生突然変異(de novo)」です。両親の検査結果によって、次のお子さんが同じ体質を持つ確率は大きく変わります。
| 親の検査結果 | 遺伝のタイプ | 次子への再発リスク |
|---|---|---|
| 両親とも欠失なし | 新生突然変異(de novo) | 低い(1%未満) ※性腺モザイクの可能性は残る |
| 片方の親が欠失あり | 家族性 | 50%(1/2) |
⚠️ 重要:親が同じ欠失を持っていても、症状が非常に軽い(または無症状)ために気づかれていないケースがあります(約25%)。そのため、次子を希望される場合は、ご両親の遺伝子検査を行うことが推奨されます。
7. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。17q12微小欠失症候群を含む染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設。NIPT後の羊水検査・絨毛検査も院内で完結し、転院の負担がありません。
💰 互助会で費用面も安心
互助会制度(8,000円)により、NIPT陽性時の確定検査費用を全額補助。追加費用の不安なく検査に進めます。
一人で悩まず、専門医を頼ってください
17q12微小欠失症候群について詳しく知りたい方、
出生前検査を検討している方は臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
🏥 一人で悩まないでください
17q12微小欠失症候群について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- [1] Mitchel MW, et al. 17q12 Recurrent Deletion Syndrome. 2016 [Updated 2020]. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. [GeneReviews]
- [2] MedlinePlus. 17q12 deletion syndrome. [MedlinePlus]
- [3] Rare Chromosome Disorder Support Group. 17q12 microdeletions. [Unique]
- [4] Orphanet. 17q12 microdeletion syndrome. [Orphanet]
- [5] Clissold RL, et al. Chromosome 17q12 microdeletions but not intragenic HNF1B mutations link developmental kidney disease and psychiatric disorder. Kidney Int. 2016;90(1):203-211. [PubMed]
- [6] Moreno-De-Luca D, et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet. 2010;87(5):618-630. [PubMed]
- [7] Roehlen N, et al. The genotypic and phenotypic spectrum of PMM2-CDG: a clinical and molecular overview. J Inherit Metab Dis. 2024. [Frontiers]
- [8] Bernardor J, et al. Phenotypic variability of 17q12 microdeletion syndrome. Clin Kidney J. 2022. [PMC]
- [9] George AM, et al. Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Reprod Biomed Online. 2021. [PubMed]
関連記事






