目次

Q. 17p13.3微小欠失症候群とはどのような病気ですか?
A. 17番染色体短腕(17p13.3)の遺伝子欠失によって生じる神経発達疾患です。
欠失の範囲によりMiller-Dieker症候群(MDS)、遠位17p13.3微小欠失症候群、孤発性滑脳症(ILS)に分類され、症状の重症度は大きく異なります。
-
➤
MDS(重症型) → 滑脳症・重度知的障害・難治性てんかん、予後不良(多くは2〜3歳で死亡) -
➤
遠位型(軽症型) → 滑脳症なし・相対的大頭症・著しい低身長・白質脳症、予後比較的良好 -
➤
原因遺伝子 → PAFAH1B1(LIS1)・YWHAE・CRKのハプロ不全 -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
頻度 → MDSは約10万出生に1例、遠位型は100万人に1人未満
1. 17p13.3微小欠失症候群とは|基本情報
【結論】 17p13.3微小欠失症候群は、17番染色体短腕末端(17p13.3)の遺伝子欠失によって生じる神経発達疾患です。欠失の範囲と含まれる遺伝子により、重症型のMiller-Dieker症候群から軽症型の遠位欠失症候群まで、症状は大きく異なります。
「検査で17p13.3欠失が見つかった」「お子さんに滑脳症がある」という方は、この病気について正確な情報を知ることが大切です。欠失の範囲によって予後が大きく異なるため、どのタイプの欠失かを正確に診断することが重要です。

17p13.3領域の17番染色体上の位置
💡 用語解説:「滑脳症」とは?
滑脳症(lissencephaly)とは、胎児期に神経細胞が正常に移動できなかったことで、大脳皮質の表面に通常あるべき脳回(しわ)が形成されず、脳が滑らかな状態になる重篤な脳形成異常です。重度の知的障害、難治性てんかん、筋緊張低下などを引き起こします。
17p13.3微小欠失症候群の3つのタイプ
17p13.3領域の欠失は、その範囲と含まれる遺伝子により3つのタイプに分類されます。最も重要なのはPAFAH1B1(LIS1)遺伝子を含むかどうかです。
| タイプ | 欠失遺伝子 | 主な症状 | 予後 |
|---|---|---|---|
| Miller-Dieker症候群(MDS) | PAFAH1B1+YWHAEを含む広範な欠失 | 滑脳症、小頭症、重度知的障害、難治性てんかん、特徴的顔貌 | 不良(多くは2〜3歳) |
| 遠位17p13.3微小欠失症候群 | YWHAE・CRK(PAFAH1B1を含まない) | 白質脳症、相対的大頭症、著しい低身長、軽度〜中等度知的障害 | 比較的良好(成人例あり) |
| 孤発性滑脳症(ILS) | PAFAH1B1のみの限局的欠失 | 滑脳症、知的障害、てんかん(顔貌異常は軽度) | 中等度 |
⚠️ 最重要ポイント:PAFAH1B1遺伝子の有無が予後を決める
PAFAH1B1(LIS1)遺伝子は神経細胞の移動に必須です。この遺伝子を含む欠失では滑脳症が生じ、予後は不良です。一方、PAFAH1B1を含まない遠位欠失では滑脳症は生じず、予後は比較的良好です。検査結果を見る際は、どの遺伝子が欠失範囲に含まれるかを確認することが極めて重要です。
疾患の頻度
| タイプ | 推定発生頻度 |
|---|---|
| Miller-Dieker症候群(MDS) | 約10万出生に1例 |
| 遠位17p13.3微小欠失症候群 | 100万人に1人未満(極めて稀) |
2. 17p13.3微小欠失症候群の主な症状
【結論】 本症候群の症状は欠失のタイプにより大きく異なります。Miller-Dieker症候群では滑脳症を中心とした重篤な症状を呈し、遠位欠失症候群では滑脳症を伴わない白質異常と成長障害が特徴です。
Miller-Dieker症候群(MDS)の症状
MDSは最も重症型で、PAFAH1B1とYWHAEの両方を含む広範な欠失により生じます。
-
•
滑脳症:大脳皮質の脳回が欠如し、脳表面が滑らかになる重篤な脳形成異常
-
•
小頭症:頭囲が著しく小さい
-
•
重度知的障害:発達の遅れが顕著
-
•
難治性てんかん:乳児期早期から発症、薬物抵抗性
-
•
筋緊張低下:哺乳障害、誤嚥のリスク
👤 MDSの特徴的顔貌
- •
前額部の突出
- •
中顔面低形成
- •
小さく上向いた鼻
- •
低位耳介
- •
小顎、厚い上口唇
🫀 MDSの合併症
- •
先天性心疾患
- •
腎奇形
- •
臍帯ヘルニア
- •
消化器奇形
遠位17p13.3微小欠失症候群の症状
遠位型はPAFAH1B1を含まないため、MDSとは全く異なる臨床像を呈します。滑脳症は生じません。
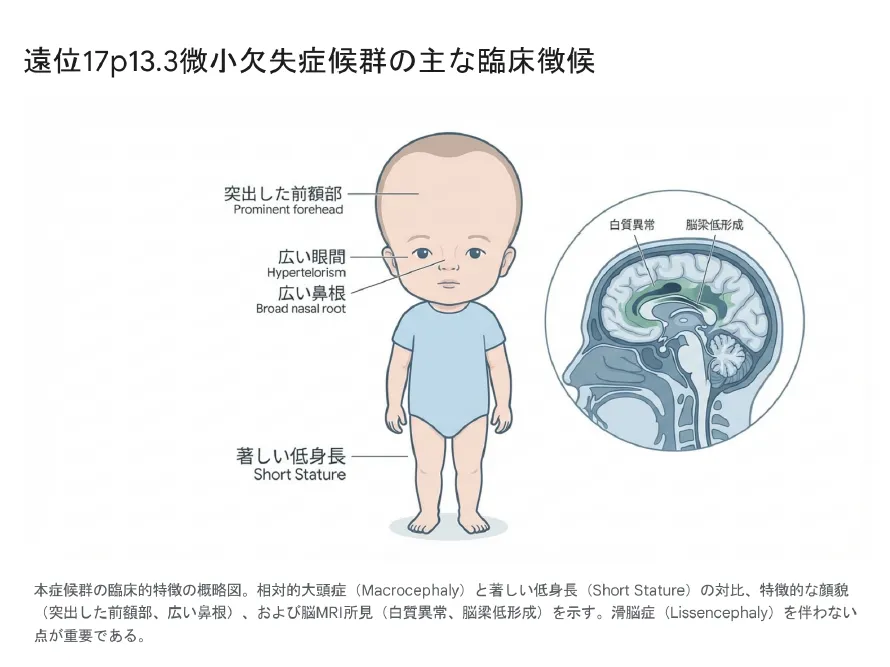
遠位17p13.3微小欠失症候群の主な臨床徴候:相対的大頭症と著しい低身長が特徴
-
•
滑脳症なし:MDSとの最大の鑑別点
-
•
白質脳症:大脳白質の信号異常(MRIで高信号)
-
•
脳梁低形成:脳梁の形成不全
-
•
Chiari I型奇形:小脳扁桃の下方偏位
-
•
軽度〜中等度知的障害:言語発達遅延が顕著
📏 遠位型の成長障害
- •
著しい低身長(-2SD〜-3SD以下)
- •
子宮内発育遅延(IUGR)
- •
相対的大頭症(低身長に対して頭囲は正常〜大きめ)
👤 遠位型の顔貌特徴
- •
突出した前額部
- •
眼間開離
- •
広い鼻根
- •
厚い口唇
💡 MDSと遠位型の決定的な違い:頭囲
MDS=小頭症、遠位型=相対的大頭症という頭囲の違いは、臨床診断における重要な手がかりです。低身長に対して頭囲が大きめに保たれている場合は遠位型を疑います。

🩺 院長コラム【欠失範囲の確認が最重要】
17p13.3欠失と診断された場合、まず確認すべきは「PAFAH1B1遺伝子が欠失範囲に含まれているか」です。この遺伝子の有無で予後は天と地ほど違います。
MDSであれば滑脳症による重篤な神経障害は避けられません。一方、遠位型であれば成人まで生存し、ある程度の自立した生活が期待できる症例も報告されています。
臨床遺伝専門医として、検査結果の正確な解釈と、それに基づく適切な予後説明を行うことが私の使命だと考えています。
3. 原因と遺伝的背景|3つの責任遺伝子
【結論】 本症候群の原因は、17p13.3領域に位置するPAFAH1B1(LIS1)・YWHAE・CRKの3遺伝子のハプロ不全です。どの遺伝子が欠失に含まれるかで臨床像が決まります。約80〜90%は新生突然変異ですが、10〜20%は親の均衡型転座に由来します。
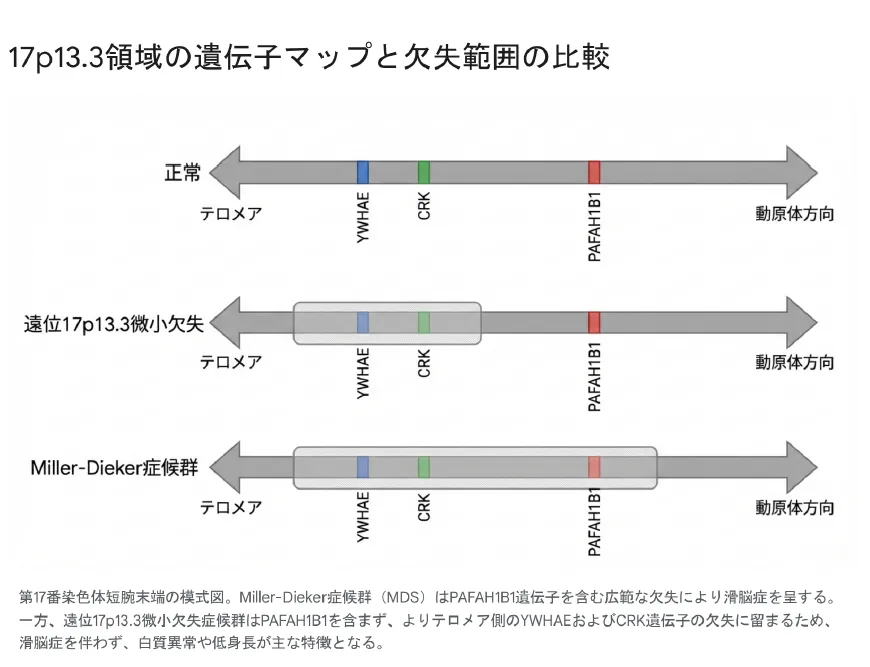
17p13.3領域の遺伝子マップと欠失範囲の比較:MDSと遠位型の違い
💡 用語解説:「ハプロ不全」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。「ハプロ不全」とは、1コピーが欠失または機能しなくなることで、残り1コピーだけでは正常な機能を維持できない状態を指します。
3つの責任遺伝子の機能
| 遺伝子 | 主な機能 | 欠失による影響 |
|---|---|---|
| PAFAH1B1(LIS1) | 神経細胞遊走に必須。微小管ダイニン複合体の調節 | 滑脳症(この遺伝子の欠失がMDSの核心) |
| YWHAE(14-3-3ε) | NDEL1-LIS1経路の調節、シナプス機能 | 白質脳症、神経細胞遊走の軽微な障害、MDSの重症度を増加 |
| CRK | IGF-1Rシグナル経路、Reelinシグナル経路 | 著しい低身長(成長ホルモンシグナル伝達障害) |
PAFAH1B1(LIS1):滑脳症の原因遺伝子
PAFAH1B1遺伝子は神経細胞遊走の最重要因子です。胎児期の脳では、神経細胞が生まれた場所から大脳皮質の適切な位置へ移動する必要があります。この移動にPAFAH1B1タンパク質は不可欠です。
-
①
微小管の安定化:細胞骨格の維持に関与
-
②
ダイニン複合体の調節:神経細胞の移動に必要なモーター機能
-
③
ハプロ不全→神経細胞遊走障害→滑脳症
YWHAE(14-3-3ε):MDSの重症度修飾因子
YWHAE遺伝子は14-3-3εタンパク質をコードし、NDEL1を介してLIS1経路を調節します。YWHAEの欠失は単独では滑脳症を引き起こしませんが、LIS1欠失と組み合わさるとMDSの重症度を増加させます。
💡 遠位欠失症候群の病態
遠位型ではPAFAH1B1は正常に存在するため、神経細胞遊走の重篤な障害(滑脳症)は生じません。しかしYWHAEの欠失により、白質の形成不全や脳構造の微細な異常が生じます。これがマウスモデルでも確認されている白質脳症の病態基盤です。
CRK:成長障害の原因遺伝子
CRK遺伝子はIGF-1R(インスリン様成長因子1受容体)シグナル経路に関与し、遠位欠失症候群における著しい低身長の主因と考えられています。
💡 CRK欠失と成長障害のメカニズム
CRK欠失患者では、血中の成長ホルモン(GH)やIGF-1の濃度が正常範囲内であっても、細胞レベルでのシグナル伝達効率が低下しているため、重度の成長不全を呈します。これが外因性のGH投与で部分的に改善できる可能性の根拠となっています。
欠失の発生機序と遺伝形式
17p13.3領域には低コピー反復配列(LCR)が存在し、減数分裂時に非対立遺伝子間相同組換え(NAHR)を誘発することで欠失が生じやすくなっています。
新生突然変異(80〜90%)
両親は正常な核型で、患児で新たに発生した欠失。次子への再発リスクは1%未満(生殖細胞モザイクの可能性を除く)。
親の均衡型転座由来(10〜20%)
片親が均衡型転座の保因者。親自身は無症状でも、配偶子形成時に不均衡転座が生じ、子に欠失が伝わる。次子への再発リスクは高い。
⚠️ 重要:本症候群は常染色体優性(顕性)遺伝形式をとります。すなわち、17番染色体の片方に欠失があれば発症します。親の検査(核型分析)により、新生突然変異か転座由来かを判別することが再発リスク評価に不可欠です。
4. 17p13.3微小欠失症候群の診断方法
【結論】 本症候群の確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のG分染法では微小欠失を検出できません。臨床的に滑脳症や特徴的顔貌が疑われた場合に検査が行われます。
診断のきっかけ
-
①
胎児超音波で脳奇形:脳溝の消失、脳室拡大などが示唆された場合
-
②
新生児のMRIで滑脳症:大脳皮質の平滑化が確認された場合
-
③
特徴的顔貌+発達遅滞:MDSを疑う臨床所見がある場合
-
④
著しい低身長+相対的大頭症:遠位型を疑う所見がある場合
-
⑤
原因不明のてんかん+知的障害:包括的遺伝学的検査として
遺伝学的検査の種類
| 検査方法 | 特徴 | 17p13.3欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。数十kb〜数Mbの微細CNVを高解像度で検出 | ◎ 検出可能 |
| G分染法(核型分析) | 解像度は5〜10Mb程度。大きな転座の検出に有用 | ✕ 検出困難(微小欠失は検出限界以下) |
| FISH法 | LIS1領域の特異的プローブで欠失を可視化 | ○ 検出可能(確認検査として有用) |
| MLPA法 | 特定領域のコピー数を定量。比較的安価 | △ 専用キットで可能 |
💡 用語解説:染色体マイクロアレイ(CMA)とは?
CMAは、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。発達遅滞や先天異常の原因検索として保険適用検査となっています。欠失の正確な範囲と含まれる遺伝子を同定できるため、MDSと遠位型の鑑別に必須です。
診断後の追加検査
17p13.3欠失が確認されたら、以下の追加検査が推奨されます。
🧠 画像検査
- •
頭部MRI:滑脳症、白質異常、脳梁形成の評価
- •
脳波検査:てんかんの評価
🫀 合併症検索
- •
心エコー:先天性心疾患の評価
- •
腹部エコー:腎奇形の評価
- •
眼科検査、聴力検査
👪 家族検査
- •
両親の核型分析:均衡型転座の有無
- •
FISH検査:欠失の由来確認
5. 治療と長期管理
【結論】 本症候群には根本的な治療法は存在せず、症状に応じた対症療法・早期療育・包括的ケアが中心となります。特に遠位型では成長ホルモン療法の有効性を示す報告もあります。
Miller-Dieker症候群(MDS)の管理
MDSは予後不良であり、緩和ケアと合併症管理が治療の主軸となります。
⚡ てんかん管理
- •
抗てんかん薬による発作コントロール
- •
多くは難治性のため複数薬剤が必要
- •
定期的な脳波モニタリング
🍼 栄養・呼吸管理
- •
経管栄養・胃瘻造設の検討
- •
誤嚥性肺炎の予防
- •
気道確保・呼吸管理
遠位型の管理:成長ホルモン療法の可能性
遠位型では予後が比較的良好であり、成長ホルモン(GH)療法による低身長改善の報告があります。
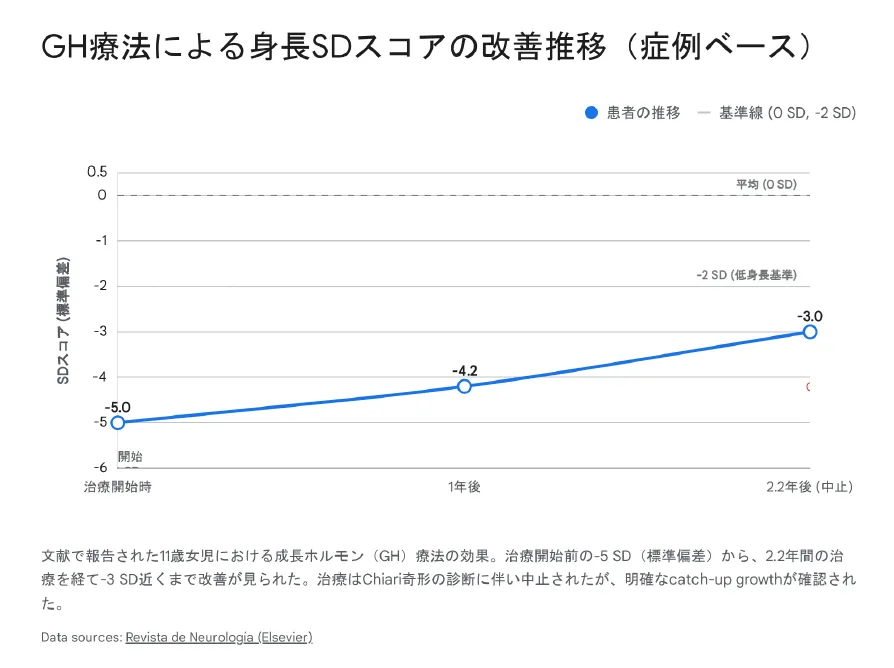
成長ホルモン療法による身長SDスコアの改善推移(症例報告より)
-
•
症例報告:11歳女児で治療前-5SD→2.2年後-3SDに改善
-
•
メカニズム:CRK欠失による末梢シグナル障害を外因性GHで部分的に代償
-
•
注意点:Chiari I型奇形合併例では頭蓋内圧亢進リスクあり、MRIモニタリング必須
ライフステージ別の管理
| ライフステージ | MDS | 遠位型 |
|---|---|---|
| 乳児期 | 栄養管理、呼吸管理、てんかん治療開始 | 早期療育開始、心エコー、発達評価 |
| 幼児期 | 緩和ケア、感覚刺激、家族支援 | PT・OT・ST、GH療法検討、特別支援教育準備 |
| 学童期〜成人期 | (多くは幼少期を超えて生存できない) | 教育支援、就労支援、移行期医療、精神的サポート |
-
•
臨床遺伝科:遺伝カウンセリング、家族への説明
-
•
小児神経科:てんかん管理、神経学的評価
-
•
リハビリテーション科:PT・OT・ST(理学療法・作業療法・言語療法)
-
•
小児循環器科:心疾患の評価・フォロー
-
•
内分泌科:成長ホルモン療法の検討(遠位型)
6. 遺伝カウンセリングの重要性
【結論】 17p13.3欠失では欠失範囲の正確な把握と親の検査による由来確認が極めて重要です。MDSと遠位型で予後が大きく異なるため、遺伝カウンセリングで正確な情報を提供することが不可欠です。
遺伝カウンセリングで伝えるべきポイント
-
①
欠失範囲の確認:PAFAH1B1を含むか否かで予後は天と地の差
-
②
親の検査:新生突然変異か転座由来かで再発リスクが大きく異なる
-
③
予後の説明:MDSは予後不良、遠位型は成人例あり
-
④
治療オプション:対症療法、早期療育、遠位型ではGH療法の可能性
-
⑤
心理的サポート:診断後の家族への継続的支援
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常核型(新生突然変異) | 1%未満(生殖細胞モザイクの可能性はあり) |
| 片親が均衡型転座保因者 | 有意に上昇(転座の種類により異なる、要専門評価) |
⚠️ 重要:約10〜20%の症例では親の均衡型転座に由来します。この場合、同一家系から複数の患児が生まれる可能性があります。再発リスク評価のため、両親の核型分析は必須です。
🩺 院長コラム【遺伝カウンセリングで大切にしていること】
17p13.3欠失症候群の遺伝カウンセリングで最も重要なのは、正確な情報提供と家族の意思決定支援です。MDSと遠位型では予後が全く異なるため、まず検査結果の正確な解釈が不可欠です。
私が臨床遺伝専門医として30年以上、のべ10万人以上のご家族の意思決定と向き合ってきた経験から言えるのは、「正しい情報を持つことで、ご家族は自分たちに合った決断ができる」ということです。
出生前診断で見つかった場合、妊娠継続の判断を迫られることもあります。どのような決断をされても、私はご家族に寄り添い続けます。一人で悩まず、ぜひご相談ください。
7. 出生前診断について|NIPTと羊水検査
【結論】 17p13.3欠失は出生前診断で検出可能です。胎児超音波で脳奇形が疑われた場合に羊水検査でのCMAが行われます。確定診断後は欠失範囲の正確な把握と遺伝カウンセリングが重要です。
出生前検査での検出方法
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| 胎児超音波 | △ 間接的 | 脳溝の消失、脳室拡大、小頭症などの所見で疑う |
| 羊水検査+CMA | ◎ 検出可能 | 確定診断のゴールドスタンダード。欠失範囲も同定可能 |
| 絨毛検査+CMA | ◎ 検出可能 | 妊娠初期(11〜14週)に実施可能 |
| 羊水検査+FISH | ○ 検出可能 | LIS1領域の欠失を直接可視化、迅速な確認に有用 |
⚠️ NIPTでの検出について
NIPTは21/18/13トリソミーのスクリーニングを主目的としており、17p13.3の微小欠失は標準検査項目に含まれていません。当院で実施しているCOATE法を用いた全染色体検査でも、この領域の微小欠失の検出は困難です。胎児超音波で脳奇形が疑われた場合は、羊水検査での確定診断が必要です。
出生前診断で見つかった場合の対応
-
①
欠失範囲の確認:PAFAH1B1を含むか(MDS)か、含まないか(遠位型)を明確に
-
②
遺伝カウンセリング:予後、治療オプション、再発リスクの説明
-
③
両親の検査:均衡型転座の有無を確認(次子への再発リスク評価)
-
④
出生後管理計画:周産期管理、出生後の検査・療育計画の準備
⚠️ 重要:出生前診断で17p13.3欠失が見つかった場合、欠失範囲により予後が大きく異なります。MDSであれば滑脳症による重篤な神経障害は避けられませんが、遠位型であれば比較的良好な予後が期待できます。正確な情報に基づいた意思決定のため、臨床遺伝専門医による遺伝カウンセリングを受けることを強くお勧めします。
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。全染色体検査や12種類の微小欠失検査に対応しています。
💰 互助会制度で費用面も安心
互助会制度(8,000円)により、陽性時の確定検査(羊水検査)費用を全額補助。NIPT受検者全員に適用されます。
一人で悩まず、専門医を頼ってください
17p13.3欠失症候群について詳しく知りたい方、
検査結果の解釈に困っている方は臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
🏥 一人で悩まないでください
17p13.3欠失症候群について心配なこと、検査結果の解釈に困っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- [1] Distal 17p13.3 microdeletion syndrome – Orphanet. [Orphanet]
- [2] 17p13.3 microdeletions – Unique (Rare Chromosome Disorder Support Group). [RareChromo.org]
- [3] Miller-Dieker syndrome – MedlinePlus Genetics. [MedlinePlus]
- [4] Chromosome 17p13.3 microdeletion syndrome with unaltered PAFAH1B1 gene. Neurologia (English Edition). 2019;34(3):208-210. [Elsevier]
- [5] Expression analysis of a 17p terminal deletion, including YWHAE, but not PAFAH1B1, associated with normal brain structure on MRI. Am J Med Genet A. 2014;164A(1):72-77. [Ovid]
- [6] Further expansion and confirmation of phenotype in rare loss of YWHAE gene distinct from Miller-Dieker syndrome. Am J Med Genet A. 2023;191(5):1325-1333. [PubMed Central]
- [7] Neurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3. Front Genet. 2018;9:80. [Frontiers]
- [8] 17p13.3 Microdeletion: Insights on Genotype-Phenotype Correlation. Mol Syndromol. 2017;8(1):22-27. [PubMed Central]
- [9] YWHAE gene – MedlinePlus Genetics. [MedlinePlus]
- [10] Distal 17p13.3 microdeletion syndrome – GARD (Genetic and Rare Diseases Information Center). [GARD/NIH]
関連記事






