目次
📍 クイックナビゲーション
4p重複症候群(第4染色体短腕重複症候群、Trisomy 4p)は、第4染色体の短腕(4p)の遺伝物質が部分的または全体的に「増える」ことで発症する、極めて稀な先天性染色体異常症候群です。世界での有病率は100万人あたり1人未満と推定され、出生前・出生後の重度な成長遅延、精神運動発達遅滞、特徴的な顔貌を中心に、心臓・腎臓・骨格・生殖器など多臓器に影響が現れます。
本症候群の大きな特徴は、報告されている症例の多くが「単独の重複」ではなく、両親のいずれかが持つ均衡型転座(バランスのとれた染色体の組み換え)に由来する不均衡型転座として発症する点です。このため、診断後の遺伝カウンセリングでは両親の染色体検査と、次のお子さんへの再発リスク評価が極めて重要になります。
さらに興味深い遺伝学的現象として、第4染色体短腕の「欠失」によって生じるウォルフ・ヒルシュホーン症候群(WHS)と、本疾患(重複)の臨床症状が一部で重なる「遺伝子量感受性の双方向性」が知られています。本記事では最新の分子遺伝学的知見と臨床データをもとに、4p重複症候群の原因・症状・診断・治療・予後・遺伝カウンセリング・出生前診断について、臨床遺伝専門医の視点から包括的に解説します。
1. 4p重複症候群とは|疾患の基本情報
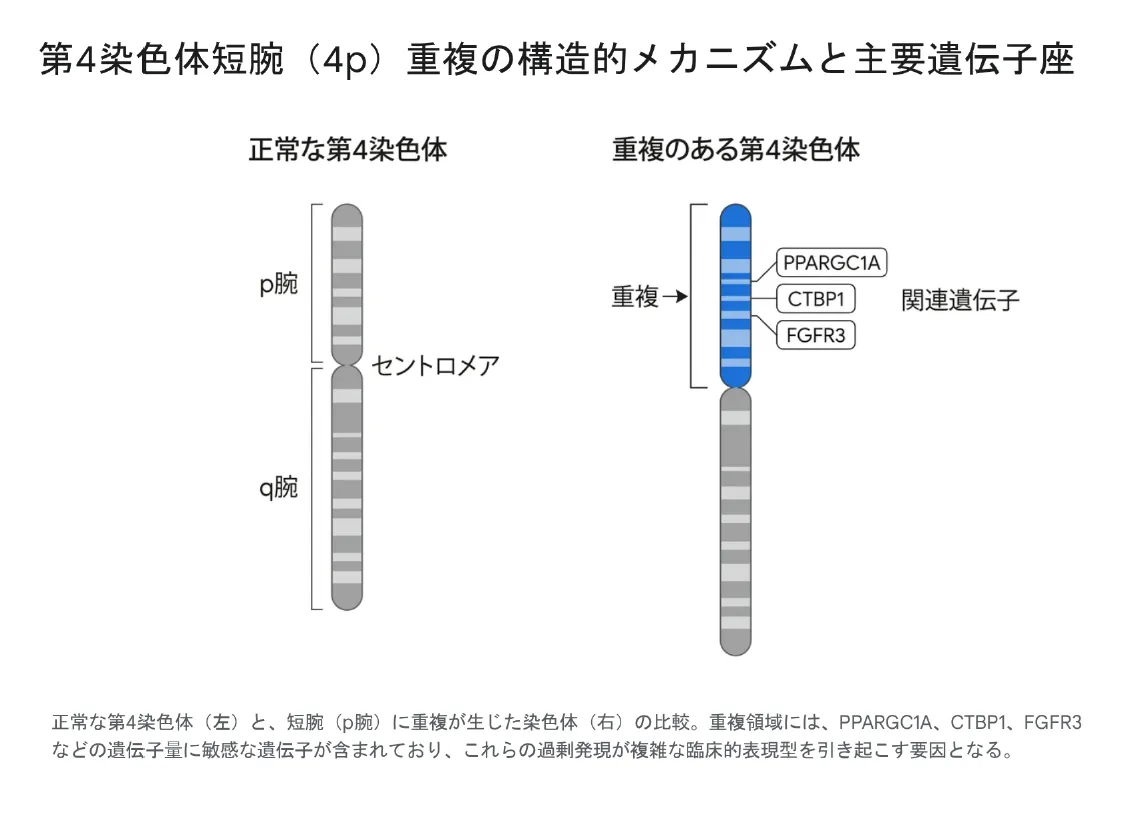
4p重複症候群は、ヒトの第4染色体の短腕(4p)の遺伝情報が、通常の2コピーから3コピー(部分的トリソミー状態)へと増えることで発症する染色体異常症候群です。第4染色体短腕は約50.86メガベース(Mb)の物理的サイズを持ち、約379の遺伝子が含まれます。これらの遺伝子の約45%が4pの遠位(テロメア側)10 Mbに高密度に集中しているため、特にこの遠位領域を含む重複では多臓器に深刻な影響が現れます。
重複している染色体セグメントの正確な物理的サイズ・切断点(ブレイクポイント)の位置・どの遺伝子が関与するかによって、軽微なものから生命を脅かす重篤なものまで臨床症状は著しく異質性が高いことが特徴です。短腕の上部3分の2以上を含む重複を持つお子さんでは、ほぼ共通する中核症状が見られます。
ヒトの体細胞は通常、各染色体を父と母から1本ずつ、計2本ずつ持っています。「トリソミー」とはある染色体(またはその一部)が3本になっている状態を指します。第4染色体全体ではなく「短腕(p腕)の一部だけ」が3本分になる場合を「部分的トリソミー4p」「4p重複」と呼びます。21トリソミー(ダウン症)のような全染色体トリソミーよりは稀ですが、影響を受ける遺伝子数が多いため重い症状が出やすいことが特徴です。
1.1 疾患の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 4p重複症候群(第4染色体短腕重複症候群、部分的トリソミー4p) |
| 英語表記 | Duplication of the short arm of chromosome 4 / Trisomy 4p syndrome |
| 原因 | 第4染色体短腕(4p)の部分的または全体的な重複 |
| 頻度 | 100万人あたり1人未満(極めて稀少) |
| 遺伝形式 | 多くは親の均衡型転座由来の不均衡型転座。新生突然変異(de novo)でも発症 |
| 主な責任遺伝子(候補) | PPARGC1A、CTBP1、TRIO、FGFR3、NSD2(旧称WHSC1)など |
| 国際分類 | ICD-10:Q92.2、ICD-11:LD41.31、Orphanet:ORPHA:1738、GARD:6091 |
1.2 ウォルフ・ヒルシュホーン症候群(4p欠失)との違い
4p重複症候群と名前が似ていてしばしば混同されるのが、第4染色体短腕の「欠失」によって起こるウォルフ・ヒルシュホーン症候群(WHS、4p-症候群)です。両者は染色体上の同じ領域に関わりますが、欠失(遺伝物質が減る)と重複(遺伝物質が増える)という正反対の遺伝子量変化が原因の異なる疾患です。
興味深いことに、4p16.3領域内のFGFR3やNSD2(WHSC1)といった遺伝子は遺伝子量感受性が双方向性であることが知られており、欠失によるハプロ不全(量が半分になること)でも、重複による過剰発現でも、脳神経系や頭蓋顔面の発達に同様に深刻な影響を与えます。そのため両疾患の臨床症状は一部で重なる「パラドックス的な表現型重複」を示し、診断には染色体マイクロアレイ検査による精密な評価が不可欠です。
1.3 単独重複と複合染色体異常
報告されている4p重複症候群の症例の大部分は、他の染色体の部分欠失(モノソミー)を伴う不均衡型転座として発症しています。特に臨床的影響が大きい組み合わせとして、第5染色体短腕末端の欠失(猫鳴き症候群、Cri-du-chat症候群)との合併があります。例えば、t(4;5)の不均衡型転座では「4p重複+5p欠失」となり、4p重複症候群の構造的奇形に加えて、猫鳴き症候群特有の高音の泣き声・重篤な発育不全が同時に現れる複合表現型を呈します。「純粋な」4p重複は非常に稀であり、診断時には他の染色体異常の合併の有無を確認することが重要です。
2. 4p重複症候群の主な症状|多系統への影響
本症候群は単一の臓器ではなく、中枢神経系・頭蓋顔面・心血管系・骨格系・泌尿生殖器系・消化器系など多系統に影響します。出生前から胎児発育に影響が現れ、出生後はほぼ全例で重度の発達遅滞・知的障害・特徴的顔貌が観察されます。
2.1 出生前超音波で検出される主要所見の頻度
📊 4p染色体異常における主要な出生前超音波所見の頻度
※ 出生前所見はコホート研究、出生後所見は文献レビューに基づく代表値
特に「鼻骨形成不全を伴う小頭症」は、4p染色体異常を含む重大な染色体異常を強く疑わせる重要な複合所見として知られています。妊娠中期の超音波スクリーニングでこれらの所見が認められた場合は、染色体異常を視野に入れた精密検査と遺伝カウンセリングへの移行が推奨されます。
2.2 中枢神経・神経発達への影響
発達遅滞は本症候群の最も普遍的な特徴であり、お子さんの自立度や生涯にわたるQOLに最も大きな影響を与えます。運動マイルストーンの達成は大幅に遅れ、寝返り・お座り・歩行開始のいずれも標準より大きく遅延します。
- 発達マイルストーン:寝返りは2〜11ヶ月、お座りは9〜19ヶ月で獲得。歩行開始は早くて24ヶ月、6歳以降になる症例も
- 知的障害:軽度から重度まで幅があるが、総じて学習には特別な支援を要する
- 言語:重度の発話遅延が見られ、言語的コミュニケーションが完全に欠如する例も稀ではない
- てんかん発作:約25%に発症。乳幼児期から見られ、厳格な医学的管理を要する
- 脳の構造異常:脳室の嚢胞性病変、脳梁欠損などの正中線形成異常が観察される
- 行動特性:基本的には明るく穏やかな性格のお子さんが多い一方、注意持続の短さ・特定の事柄への執着・睡眠障害が見られる
2.3 特徴的な頭蓋顔面形態
4p重複症候群に共通する顔貌は、複数の所見が組み合わさって独特の印象(gestalt)をつくり、臨床医が本症候群を疑う重要な手がかりとなります。これらの形態異常は、胎生期の神経堤細胞の移動や鰓弓の発達過程における遺伝子発現の乱れに起因すると考えられています。
- 頭部・前頭部:小頭症、突出した前額部、眉間部の隆起、眉毛を横断する骨の隆起、低いヘアライン
- 眼:両眼開離(hypertelorism)、斜視、まれに小眼球症や虹彩コロボーマ(虹彩の鍵穴状の裂け目)
- 鼻:広く平坦な鼻梁、特徴的な球根状の鼻尖(「ボクサーの鼻」と形容される丸み)
- 口腔:小さな口、薄い口唇、長い人中、高口蓋、口蓋裂を伴うことも
- 顎:下顎発達不全による小顎症・下顎後退、尖った顎
- 耳:大きく低い位置に付着した耳介、耳輪・対耳輪の形状異常
- 頸部:短い首
2.4 筋骨格系・四肢の特徴
運動機能の発達を妨げる重要な要因として、乳児期の重度な筋緊張低下(hypotonia、いわゆるフロッピーインファント)がほぼ全例で見られます。成長に伴って一部の患者さんでは逆に筋緊張亢進へ変化し、関節の拘縮を引き起こすことがあります。
- 脊柱:成長期の約38%に脊柱側弯症が進行し、呼吸機能や姿勢保持に影響
- 手指:蜘蛛状指(arachnodactyly:細く長い指)、屈曲指(camptodactyly:曲がったまま伸びない指)、親指の付着位置異常
- 足部:内反足(club foot)、「揺り椅子状足底(rocker bottom feet)」は本症候群を強く疑わせる特徴的所見
足底(足の裏)が外側に湾曲し、踵が後方に突出することで、横から見ると揺り椅子のような形状になる足の変形です。エドワーズ症候群(18トリソミー)でよく知られていますが、4p重複症候群でも特徴的な所見の一つで、診断の重要な手がかりとなります。
2.5 重要な内臓奇形
生命予後に直結し、継続的な医療介入を必要とする内臓奇形が複数の臓器系で認められます。
- 心血管系:心室中隔欠損(VSD)・心房中隔欠損(ASD)・大動脈縮窄症などが報告される。新生児期のチアノーゼ・呼吸障害・不整脈の原因となる
- 泌尿器系:腎低形成(renal hypoplasia)など
- 男性生殖器:極端に小さな陰茎(micropenis)、停留精巣、尿道下裂が高頻度
- 消化器系:約30%に何らかの胃腸系奇形(先天的な胆嚢欠損、約15%に鼠径ヘルニア)
- 呼吸器系:後鼻孔閉鎖症(choanal atresia)による新生児期の呼吸障害、繰り返す呼吸器感染症

3. 原因と責任遺伝子|なぜ症状が起こるのか
4p重複症候群の症状は、4p16.3〜4p13などの領域内に存在する複数の遺伝子のコピー数が2から3に増えることで生じる「遺伝子量効果(Gene Dosage Effect)」に起因します。遺伝子は通常2つの対立遺伝子から正確な量のタンパク質を産生するように制御されていますが、コピー数が増えることで特定のタンパク質が過剰に発現し、胚発生時の細胞分化・増殖・細胞骨格の形成プロセスに連鎖的な機能不全を引き起こします。
通常、私たちは父と母から1コピーずつ、計2コピーの遺伝子を受け継いでいます。「遺伝子量効果」とは、遺伝子のコピー数が変化することでその遺伝子から作られるタンパク質量も変化し、症状が現れる現象です。遺伝子量に敏感な遺伝子(dosage-sensitive gene)では、1コピーになっても(ハプロ不全)、3コピーになっても(過剰発現)、いずれも発生過程の異常を引き起こします。
3.1 主な責任遺伝子と役割
| 遺伝子 | 主な役割 | 過剰発現で関連する症状 |
|---|---|---|
| PPARGC1A | 細胞のエネルギー代謝、ミトコンドリア機能の調節 | 神経精神運動発達遅滞、小頭症 |
| CTBP1 | 転写共抑制因子、神経系発達の遺伝子発現調整 | 脳の構造的変化、神経発達障害 |
| TRIO | 細胞骨格の動態、アクチンリモデリング制御 | 神経突起の伸長・細胞移動の障害 |
| FGFR3 | 線維芽細胞増殖因子受容体、遺伝子量に非常に敏感 | 骨格異常、顔面形態の異常 |
| NSD2(旧称WHSC1) | クロマチンリモデリング、遺伝子発現の制御 | 発達障害、頭蓋顔面の発生異常 |
これらの遺伝子は単独で機能しているのではなく、細胞の分化・増殖・細胞骨格形成において中心的なハブとして相互に連携した緻密なネットワークを形成しています。重複によりコピー数が増えると、このネットワーク全体に「ノイズ」が生じ、結果として多臓器にわたる複雑な表現型を発現させます。
3.2 隣接遺伝子症候群としての性質
4p重複症候群は単一の遺伝子疾患ではなく、染色体上で隣り合って並ぶ複数の遺伝子が一度に重複する「隣接遺伝子症候群(contiguous gene syndrome)」です。それぞれの遺伝子が異なる役割を担っているため、脳・心臓・骨格・呼吸器など複数の臓器に同時に影響が出るのが特徴です。
3.3 ウォルフ・ヒルシュホーン症候群(4p欠失)とのパラドックス的な表現型重複
遺伝学的に極めて興味深い現象として、4p16.3領域の欠失で生じるウォルフ・ヒルシュホーン症候群(WHS)と、本疾患(重複)との間には臨床症状の「パラドックス的な重複」が存在します。実際に、原因不明の精神遅滞をもつ患者さんの精密検査で4p16.3領域の微細重複(microduplication)が見つかり、その臨床像がWHSの典型症状と驚くほど類似していた症例が報告されています。
これは、4p16.3領域のFGFR3やNSD2(WHSC1)といった遺伝子量感受性遺伝子が、欠失によるハプロ不全(量が半分)でも、重複による過剰発現でも、脳神経系や頭蓋顔面の発達に同様に深刻な影響を与えるという「双方向性」を持つことを示しています。WHSのような臨床所見を呈しながら核型分析で欠失が見られない患者さんに対しては、重複の可能性を疑って高解像度のマイクロアレイ検査を実施することが鑑別診断上必須です。
3.4 発症パターン|親の均衡型転座と新生突然変異
本症候群の発症メカニズムには、遺伝カウンセリングにおいて全く異なる意味合いを持つ2つのパターンがあります。
| 発症パターン | メカニズム | 次子への再発リスク |
|---|---|---|
| 親の均衡型転座由来 (多くの症例) |
片親が4番染色体と他の染色体との間に均衡型の構造的再配列を持つ。配偶子形成時の不均等分配により、不均衡型として遺伝 | 有意に上昇(転座の種類により変動、要個別評価) |
| 新生突然変異(de novo) | 両親の染色体核型は正常。配偶子形成時または受精直後の細胞分裂で突発的に組み換えエラーが発生 | 原則として低い(一般人口と同程度) |
・均衡型転座(balanced translocation):2つの染色体の一部が交換されているが、全体として遺伝情報の総量に過不足がない状態。保因者は通常、健康に成長し気づかれないことが多い。
・不均衡型転座(unbalanced translocation):遺伝情報の量に過不足が生じている状態。本症候群のように、一方の染色体の一部が「重複」し、対となるもう一方の染色体の一部が「欠失」しているケースなど。臨床症状を引き起こす。
・新生突然変異(de novo):両親には染色体異常がなく、お子さんで新たに発生した突然変異を意味する。本症候群でも一定の頻度で見られる。
親の妊娠前の生活習慣・環境要因・特定の行動が新生突然変異を引き起こすわけではないという点を、医師は家族に対して明確に伝える必要があります。これは染色体形成過程における偶発的な事故であり、ご両親の責任ではありません。
4. 4p重複症候群の診断方法と鑑別診断
4p重複症候群の診断において、現代の臨床遺伝学者が留意すべき最も重要な点は「標準的なGバンド法による核型分析だけでは微細な異常を見逃すリスクが高い」ということです。数メガベース未満の微小重複は従来の染色体検査の解像度の限界を下回るため、核型が「正常」と誤診される危険性があります。
4.1 出生後の確定診断|染色体マイクロアレイ検査がゴールドスタンダード
出生後に4p重複症候群が疑われる場合、血液検体を用いた染色体マイクロアレイ検査(CMA)がゴールドスタンダードとなります。CMAはゲノム全体にわたるコピー数変異(CNV)を高解像度で包括的にスキャンし、微小重複や微小欠失の存在・正確な物理的サイズ・切断点に含まれる遺伝子群を正確に特定します。確定診断後は両親の染色体検査を行い、新生突然変異か親由来の不均衡型転座かを判定し、頭部MRI・心エコー・腎エコー・眼科耳鼻科診察・脳波などで合併症の精査を進めます。
CMA(chromosomal microarray analysis)は、従来のGバンド法では検出できない数kb〜数Mb単位の微小な欠失や重複(コピー数変異:CNV)を網羅的に検出する検査です。日本では原因不明の発達遅滞・知的障害・多発奇形に対する保険適用検査として実施されており、4p重複症候群の確定診断には欠かせません。
4.2 検査方法ごとの違い
| 検査方法 | 特徴 | 4p重複の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 確定診断のゴールドスタンダード。微細CNVを高解像度で検出 | ◎ 確実に検出 |
| Gバンド法(核型分析) | 解像度は約5〜10Mb。大規模な重複は検出可能だが微小重複は限界 | △ 大きな重複は検出可、微小重複は見逃しリスク |
| FISH法 | 特定領域の蛍光プローブで視覚的に確認 | ○ 専用プローブで確認可能 |
| MLPA法 | 特定遺伝子(FGFR3など)のコピー数を低コストで定量化 | ○ 4p16.3領域の評価に有用 |
4.3 鑑別診断|似た症状を示す疾患
4p重複症候群は症状が多彩なため、初期評価では他の遺伝性症候群と紛らわしいことがあります。鑑別すべき疾患の代表例を以下に示します。
- ウォルフ・ヒルシュホーン症候群(WHS、4p欠失症候群):同じ4p16.3領域の「欠失」による疾患。本症候群の「重複」と表現型が一部重なる「双方向性」のため、CMAでの欠失か重複かの判定が必須
- 猫鳴き症候群(Cri-du-chat syndrome、5p欠失):不均衡型転座由来の4p重複は5p欠失を合併することが多く、両者の複合表現型として現れる
- エドワーズ症候群(18トリソミー):揺り椅子状足底・小頭症・発育不全など外見上の重なりがあるが、CMAで明確に区別される
- その他の部分的トリソミー症候群:4p以外の染色体部分重複でも類似した重度発達遅滞を示すことがあり、網羅的な染色体解析が必要
お子さんの発達や検査結果が気になっていませんか?
原因不明の発達遅滞や多発奇形には染色体マイクロアレイ検査が有効です。
臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
5. 治療と長期管理|多職種チームでの包括的サポート
4p重複症候群は染色体レベルの遺伝子疾患であり、現在の医学では根本的な治療法(遺伝子治療など)は確立されていません。臨床管理の主眼は、症状ごとの対症療法・合併症の予防・発達機能の最大化・お子さんとご家族のQOL向上に置かれます。これは単一の診療科では対応不可能であり、小児科を司令塔とした多職種による学際的チームアプローチを必要とします。
5.1 新生児期の救命対応
出生直後に最も迅速な対応が必要なのは、重度の先天性心疾患・後鼻孔閉鎖症による呼吸障害・哺乳困難です。重症例では出生前から新生児集中治療室(NICU)と小児外科を備えた高次医療機関での出産計画が望ましいとされています。
- 呼吸管理:後鼻孔閉鎖症や気道異常がある場合、人工呼吸器管理や気管挿管
- 心疾患管理:VSD・ASD・大動脈縮窄症などへの薬物療法、安定後の外科的修復
- 栄養管理:顎・口腔の形態異常と筋緊張低下による哺乳困難に対し、経鼻胃管や胃瘻造設を検討
- 誤嚥防止:増粘剤を加えたミルク、食事形態の工夫
5.2 ライフステージ別の管理
| ライフステージ | 主な対応 |
|---|---|
| 新生児期(0〜28日) | 心疾患・呼吸障害の救命管理、外科的修復、哺乳支援、合併症の精査 |
| 乳児期・幼児期(〜5歳) | 早期療育(PT・OT・ST)、口蓋裂手術、停留精巣手術、てんかんの早期管理 |
| 学童期(6〜12歳) | 特別支援教育、脊柱側弯症への装具・手術、視覚・聴覚の継続評価、歯科矯正 |
| 思春期・成人期 | 移行期医療、生活自立支援・就労支援、家族介護負担への支援 |
5.3 てんかんの管理
本症候群では約25%にてんかん発作が認められ、脳の発達をさらに後退させるリスクがあるため厳格なコントロールが求められます。一般的な抗てんかん薬による治療が第一選択ですが、難治性の発作に対してはビタミン補充療法や、脳の代謝を変化させて発作を抑制するケトン食療法、迷走神経刺激療法(VNS)などの選択肢があります。小児神経科医と連携し、定期的な脳波(EEG)モニタリングを行います。
5.4 早期療育・リハビリ・教育的介入
重度の発達遅滞・運動発達遅滞に対して、乳幼児期からの継続的な早期療育がお子さんの可能性を最大限に引き出します。
- 理学療法(PT):筋緊張低下や関節拘縮への介入、内反足のギプス固定後の運動機能獲得、水泳・ハイドロセラピー、足底装具・特殊靴の処方
- 作業療法(OT):微細運動・日常生活動作(ADL)の習得
- 言語聴覚療法(ST):言語遅延への訓練、発話困難な場合の手話・タッチスクリーン式コミュニケーションツール(AAC)の活用
- 外科的サポート:口蓋裂修復、心疾患手術、停留精巣固定術、尿道下裂修復、進行性脊柱側弯症への手術
- 多職種チーム:臨床遺伝科・小児科・小児外科・小児神経科・小児循環器科・整形外科・眼科・耳鼻科・心理職・ソーシャルワーカーが連携
5.5 長期予後について
本症候群の長期予後は、重複している染色体セグメントの物理的サイズ・セントロメアの関与の有無・致死性の心疾患や中枢神経系異常の重症度によって大きく異なります。セントロメアを巻き込むような大規模な重複や完全な三倍体を伴うケースでは、胚発生段階での流産や乳児期早期での死亡リスクが高い一方、微細重複(microduplication)など比較的小さな遺伝子領域の不均衡にとどまる軽度な症例では予後が大きく改善します。適切な多職種ケア・てんかん制御・適応行動の学習支援を通じて、社会生活に参加されるお子さんも報告されています。文献レビューでは24歳の成人患者が最年長の生存報告例として記録されており、本疾患が必ずしも小児期に限定される致死性疾患ではないことが示されています。
6. 遺伝カウンセリングと再発リスク
4p重複症候群の診断は、ご家族に深刻な心理的・感情的ストレスをもたらします。遺伝カウンセリングを通じて、ご家族が病気を正確に理解し、納得のいく決断ができるよう中立的な情報提供を行うことが、医師の重要な役割です。
6.1 カウンセリングで伝えるべきポイント
- 重複範囲と症状の関係:含まれる遺伝子・切断点の位置・他染色体異常の合併によって症状の重症度が変わる
- 表現型の多様性:軽症から致死的なものまで幅広いスペクトラムがある
- 予後の不確実性:同じ重複領域でも経過は個人ごとに異なる
- 両親の検査:新生突然変異か親の均衡型転座由来かを判定し再発リスクを評価
- 支援体制:多職種チーム、療育、社会福祉制度、家族会の紹介
6.2 再発リスクと次回妊娠への計画
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも染色体核型正常(新生突然変異) | 原則として一般人口と同程度(極めて低い)※生殖細胞モザイクの可能性は残る |
| 片親が均衡型転座保因者 | 有意に上昇(転座の種類・染色体組み合わせにより変動) |
親が均衡型転座保因者と判明した場合、次回妊娠における再発リスク評価と並行して、体外受精と着床前遺伝学的検査(PGT-SR)の選択肢、あるいは妊娠早期の絨毛検査・羊水検査による出生前診断のオプションが提供されます。これらの選択をするかどうかは、ご家族の人生観や価値観に深く関わる事柄であり、医師は十分な情報提供と心理的サポートを行ったうえで、決定はご家族に委ねるスタンスを貫きます。
7. 出生前診断とミネルバクリニックのサポート体制
4p重複症候群は、NIPTのうち全染色体スクリーニング型のプランでリスクを評価でき、羊水検査・絨毛検査でCMAを行うことで確定診断ができます。ただし、出生前に見つけることが常にご家族の利益になるとは限らないため、検査前後の遺伝カウンセリングが不可欠です。
7.1 出生前検査の種類と検出能力
| 検査 | 位置づけ | 4p重複への対応 |
|---|---|---|
| NIPT(ターゲット型/12微小欠失プラン) | スクリーニング検査 | ✕ 対象外(4p16欠失は対象だが、4p重複は対象外) |
| NIPT(全染色体WGS型) | スクリーニング検査 | ○ スクリーニング可能(5Mb以上の重複・欠失を全染色体で検出) |
| 絨毛検査+CMA | 確定診断 | ◎ 妊娠初期に確定診断可能 |
| 羊水検査+CMA | 確定診断 | ◎ 微小重複も確定診断可能 |
7.2 ミネルバクリニックのNIPTプランと4p重複症候群
ダイヤモンドプランの12微小欠失リストには「4p16欠失」(ウォルフ・ヒルシュホーン症候群)が含まれていますが、これは「欠失(遺伝物質が減る)」を見る項目です。本記事のテーマである「4p重複(遺伝物質が増える)」は正反対の遺伝子量変化であり、ダイヤモンドプランのターゲット検査では検出対象外となります。
ミネルバクリニックでは、ご家族のニーズに応じて複数のNIPTプランをご用意しています。各プランの4p重複症候群への対応は次のとおりです。
- スタンダードプラン(6か所7疾患):ウォルフ・ヒルシュホーン症候群(4p16.3欠失)は対象。4p重複症候群は対象外
- ダイヤモンドプラン(12微小欠失):1p36・4p16・5p15・22q11.2など特定12箇所の「欠失」を高い陽性的中率で検出。4p重複は対象外。なお、ダイヤモンドプランは同じ領域でコピー数が増える「重複」を副次的に検出する場合があり、その結果の意味づけは遺伝カウンセリングで詳しく説明します
- インペリアルプラン(WGS+ターゲット型):5Mb以上の全染色体微小欠失・重複を広範囲にスクリーニングするため、4p重複領域もカバー対象。陽性的中率は60〜70%台のため、陽性時は羊水検査・絨毛検査による確定診断が必要
7.3 出生前診断で見つかった場合の対応
出生前に4p重複が見つかった場合、本症候群は表現型の幅が非常に広く、胎児期の超音波所見だけでは将来の予後を正確に予測することが難しいことを理解しておく必要があります。遺伝カウンセリングで重複範囲・関与する遺伝子・表現型の幅・予後の不確実性を中立的に説明し、両親の染色体検査で新生突然変異か親の均衡型転座由来かを判定、詳細超音波で心奇形・脳の構造異常・四肢異常・腎異常などを精査します。重度の心疾患・呼吸器系合併症が疑われる場合はNICUを備えた高次医療機関での出産を検討し、ご家族の不安や葛藤に寄り添い、決断を急がせない時間と環境を確保することが何より大切です。
⚖️ 倫理的なスタンス|検査は「常に利益」ではない
本症候群のように不完全浸透や表現型の幅が大きい疾患では、出生前に見つけたことが必ずしもご家族の利益になるとは限りません。「特定の検査を勧める」「安心を保証する」「不安をあおる」ような表現は適切ではないと私たちは考えています。検査を受けるかどうか、結果をどう受け止めるかは、十分な情報を得たうえで、ご家族自身が決めるべき事柄です。
7.4 ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。4p重複症候群を含む染色体微小重複・微小欠失症候群について、出生前検査から結果説明、確定検査、その後のフォローまで一貫してサポートいたします。
- 全染色体スクリーニング対応:インペリアルプランでは5Mb以上の全染色体微小欠失・重複を広くスクリーニング、4p重複領域もカバー対象
- 確定検査も院内で実施:羊水検査・絨毛検査も院内で実施可能、転院の必要なし
- 臨床遺伝専門医が担当:臨床遺伝専門医が検査前後の遺伝カウンセリングを直接担当
- 互助会で費用面も安心:NIPT受検者全員に適用される互助会(8,000円)により、陽性時の羊水検査費用が全額補助されます
- 検査技術:COATE法などの高精度技術により、微細な染色体変化も丁寧に評価
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
よくある質問(FAQ)
関連記事
参考文献
- Orphanet – Trisomy 4p (ORPHA:1738) [外部サイトへ]
- GARD – Trisomy 4p (GARD:6091) [外部サイトへ]
- Unique – Duplications of 4p factsheet (Rare Chromosome Disorder Support Group) [外部サイトへ]
- Patel SV et al. Clinical manifestation of trisomy 4p syndrome. Eur J Pediatr. 1995 [外部サイトへ]
- Centerwall WR, Beatty-DeSana JW. The trisomy 4p syndrome. Am J Med Genet. 1975 [外部サイトへ]
- Genotype-phenotype correlation of deletions and duplications of 4p: case reports and literature review. Front Genet. 2023 [外部サイトへ]
- Candidate Genes Associated With Neurological Findings in a Patient With Trisomy 4p16.3 and Monosomy 5p15.2. PMC. 2020 [外部サイトへ]
- A large Indian family with rearrangement of chromosome 4p16 and 3p26.3 and divergent clinical presentations. Am J Med Genet A. 2015 [外部サイトへ]
- Prenatal sonographic findings in confirmed cases of Wolf-Hirschhorn syndrome. PMC. 2022 [外部サイトへ]
- Prenatal detection of a de novo terminal inverted duplication 4p in a fetus with the Wolf-Hirschhorn syndrome phenotype. Prenat Diagn. 2005 [外部サイトへ]
- Inv dup del(4)(:p13→p16.3::p16.3→qter) in a Girl Without Typical Manifestations of Wolf-Hirschhorn Syndrome. Am J Med Genet A. 2009 [外部サイトへ]
- CLINICAL MANIFESTATIONS OF PARTIAL TRISOMY 4p. Balkan Journal of Medical Genetics [外部サイトへ]
- Trisomy 4p – Chromosome Disorder Outreach [外部サイトへ]
- The new Wolf-Hirschhorn syndrome critical region (WHSCR-2): A description of a second case. Am J Med Genet A. 2005 [外部サイトへ]







