目次
ルビンシュタイン・テイビ症候群とは?
原因・症状・診断・検査を臨床遺伝専門医が解説
Q. ルビンシュタイン・テイビ症候群とはどのような病気ですか?
A. 幅広く角度のついた母指・母趾、特徴的な顔貌、中等度〜重度の知的障害を主な特徴とする先天異常症候群です。
CREBBP遺伝子(55〜60%)またはEP300遺伝子(8〜10%)の変異が原因で、ヒストンアセチル化の異常による「クロマチノパチー」として分類されます。
-
➤
原因遺伝子 → CREBBP(55〜60%)、EP300(8〜10%)、約30%は原因不明 -
➤
三大徴候 → 幅広い母指・母趾、特徴的顔貌(下垂した鼻中隔、しかめ笑い)、知的障害(平均IQ 35〜50) -
➤
重要な注意点 → 麻酔リスクが高い(気道確保困難、不整脈リスク)、ケロイド形成傾向 -
➤
診断方法 → 臨床診断基準(2024年国際コンセンサス)+遺伝学的検査で確定 -
➤
頻度 → 出生10万〜12.5万人に1人(稀な疾患)
1. ルビンシュタイン・テイビ症候群とは|基本情報
【結論】 ルビンシュタイン・テイビ症候群(Rubinstein-Taybi Syndrome:RSTS)は、幅広く角度のついた母指・母趾、特徴的な顔貌、知的障害を三大徴候とする先天異常症候群です。1963年にRubinsteinとTaybiが報告し、現在では「クロマチノパチー(クロマチン異常症)」として分類される、エピジェネティクス疾患の代表格です。
「お子さんがルビンシュタイン・テイビ症候群と診断された」「検査で遺伝子変異が見つかった」という方は、この病気の特徴と適切な管理について正確な情報を知ることが大切です。本症候群は多系統にわたる症状を呈しますが、90%以上が成人まで生存し、適切な支援により社会生活を送ることが可能です。
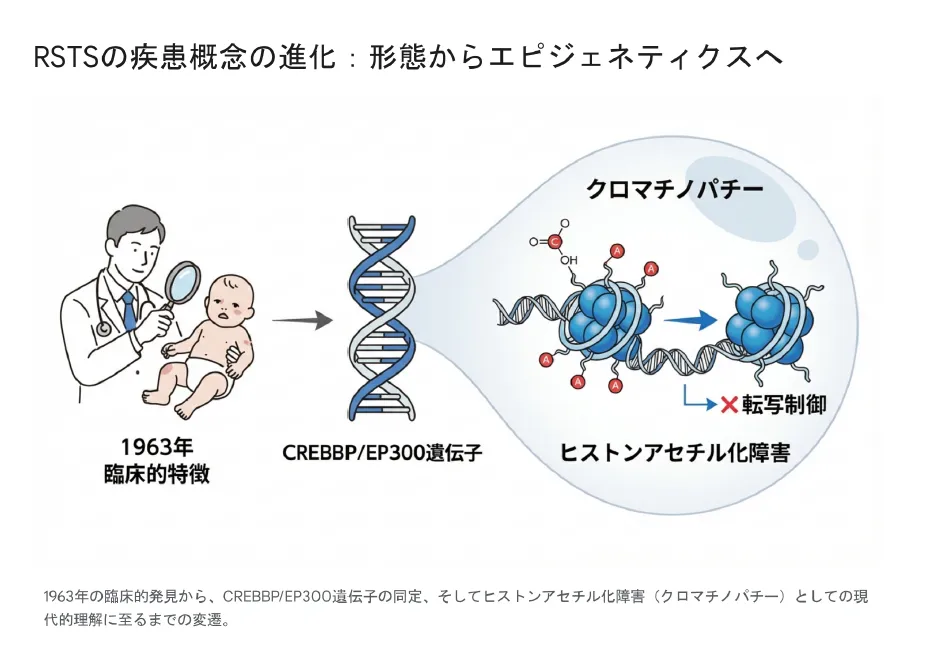
▲ RSTSの疾患概念の変遷:1963年の臨床的発見から、遺伝子同定、そして現在のクロマチノパチーとしての理解へ
💡 用語解説:「クロマチノパチー」とは?
クロマチノパチーとは、クロマチン(DNAとヒストンタンパク質の複合体)の構造や修飾に異常が生じることで発症する疾患群です。RSTSの原因遺伝子CREBBP/EP300はヒストンアセチルトランスフェラーゼ(HAT)活性を持ち、この機能低下により遺伝子発現調節が乱れ、多様な症状が生じます。
ルビンシュタイン・テイビ症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | ルビンシュタイン・テイビ症候群(OMIM #180849, #613684) |
| 別名 | 広母指・母趾症候群(Broad Thumb-Hallux Syndrome) |
| 原因 | CREBBP(55〜60%)、EP300(8〜10%)、約30%は原因不明 |
| 頻度 | 出生10万〜12.5万人に1人 |
| 遺伝形式 | 常染色体優性(顕性)遺伝、ほとんどが新生突然変異 |
| 染色体座位 | CREBBP: 16p13.3 / EP300: 22q13.2 |
疾患概念の変遷
ルビンシュタイン・テイビ症候群の理解は、この60年で大きく進歩しました。
1963年:臨床的発見
RubinsteinとTaybiが「幅広い母指・母趾、顔貌異常、知的障害」を特徴とする症候群として報告。臨床的特徴に基づく診断の時代が始まりました。
1995年:CREBBP遺伝子同定
16p13.3に位置するCREBBP遺伝子が原因遺伝子として同定され、遺伝学的診断が可能になりました。
2005年:EP300遺伝子同定
22q13.2に位置するEP300遺伝子が第二の原因遺伝子として同定。遺伝的異質性が明らかになりました。
現在:クロマチノパチーとして
両遺伝子がHAT活性を持つことから、エピジェネティクス異常による疾患として理解されています。
2. 原因遺伝子と遺伝形式
【結論】 本症候群の原因はCREBBP遺伝子(55〜60%)またはEP300遺伝子(8〜10%)の変異です。両遺伝子はともにヒストンアセチルトランスフェラーゼ(HAT)活性を持ち、遺伝子発現のエピジェネティック制御に重要な役割を果たしています。
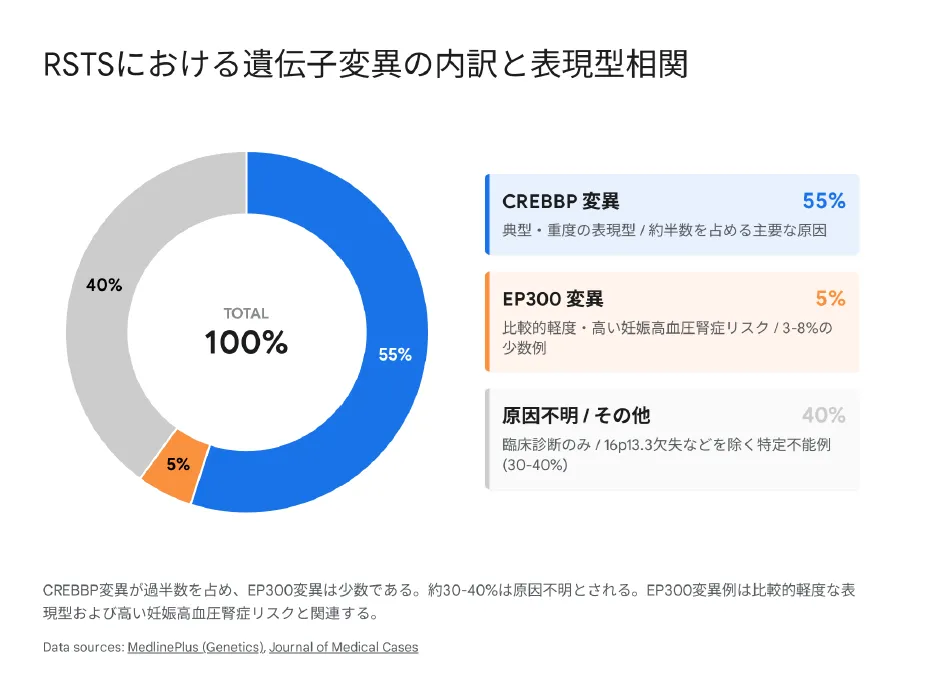
▲ RSTS原因遺伝子の内訳:CREBBP変異が過半数を占め、EP300変異は約10%、約30%は原因不明
CREBBP遺伝子とEP300遺伝子
| 遺伝子 | 染色体座位 | 頻度 | 主な機能 |
|---|---|---|---|
| CREBBP | 16p13.3 | 55〜60% | HAT活性、転写共役因子、CREB結合 |
| EP300 | 22q13.2 | 8〜10% | HAT活性、転写共役因子、CREBBPと機能相同 |
| 不明 | - | 約30% | 臨床的にRSTSだが遺伝子変異未同定 |
💡 用語解説:「ハプロ不全」とは?
RSTSはハプロ不全という機序で発症します。通常、遺伝子は父母から1本ずつ、計2コピー持っています。RSTSでは1コピーが機能しなくなることで、タンパク質産生量が50%に低下し、正常な機能を維持できなくなります。HAT活性の低下により、多くの遺伝子のアセチル化が障害されます。
遺伝子型-表現型相関:CREBBP vs EP300
EP300変異によるRSTSは、CREBBP変異と比較して全般的に軽症であることが知られています。
CREBBP変異(典型的RSTS)
- •
典型的な顔貌所見
- •
母指の角度形成が顕著
- •
知的障害は中等度〜重度
- •
ケロイド形成が多い
EP300変異(軽症型)
- •
顔貌所見は軽度〜非典型的
- •
母指は幅広いが角度形成稀
- •
知的障害は軽度〜正常のことも
- •
母体子癇前症リスク高い(23% vs 3%)
⚠️ 重要:EP300変異と妊娠合併症
EP300変異を持つ胎児を妊娠した場合、母体の子癇前症リスクが有意に高い(23% vs CREBBP 3%)ことが報告されています。また、子宮内発育不全(IUGR)の頻度も高いため、妊娠管理には注意が必要です。
遺伝形式と発生機序
RSTSは常染色体優性(顕性)遺伝ですが、ほとんどの症例は新生突然変異(de novo)として発生します。
-
①
点変異・小欠失/挿入:約60〜70%(シークエンス解析で検出)
-
②
大きな欠失/重複:約10〜15%(MLPA、CMAで検出)
-
③
モザイク:稀だが存在(体細胞モザイクの場合、軽症のことが多い)
3. 主な症状|臨床的特徴
【結論】 RSTSの三大徴候は①幅広く角度のついた母指・母趾、②特徴的顔貌、③知的障害です。多系統にわたる症状を呈し、先天性心疾患(33%)、停留精巣(男性ほぼ100%)、消化器症状、眼科的異常など、包括的な管理が必要です。
三大徴候の詳細
👍 幅広い母指・母趾
- •
橈側への角度形成が特徴的
- •
末節骨の幅広さ
- •
X線で「デルタ骨端(三角形骨端)」
- •
他の指趾も幅広いことがある
😊 特徴的顔貌
- •
下外方へ傾斜した眼瞼裂
- •
下垂した鼻中隔(low-hanging columella)
- •
しかめ笑い(grimacing smile)
- •
高口蓋、弓状眉毛
🧠 知的障害
- •
平均IQ:35〜50(中等度〜重度)
- •
言語発達の遅れ
- •
EP300変異では軽度〜正常も
- •
友好的で社交的な性格
主な臨床症状の頻度
| 症状カテゴリー | 頻度 | 詳細 |
|---|---|---|
| 成長障害 | 73% | 出生後成長不全、小頭症、成人身長:男性約162cm、女性約151cm |
| 眼科的異常 | 約80% | 斜視、屈折異常、涙道閉塞、緑内障 |
| 先天性心疾患 | 約33% | 心房中隔欠損、心室中隔欠損、動脈管開存、大動脈縮窄など |
| 停留精巣(男性) | ほぼ100% | 両側性が多い、早期手術が必要 |
| 腎尿路異常 | 27% | 水腎症、重複腎盂尿管、膀胱尿管逆流 |
| 消化器症状 | 88% | GERD、重度便秘、哺乳困難 |
| 行動特性 | 多数 | 友好的性格、ADHD様症状、ASD特性(41%)、不安(27〜64%) |
歯科的特徴:タロンカスプ
🦷 タロンカスプ(Talon Cusp)とは?
タロンカスプは、上顎切歯の舌側(内側)に生じる爪状の副咬頭です。RSTSに高い特異度を持つ所見であり、この所見があればRSTSを強く疑う根拠となります。永久歯に多く、う蝕や咬合異常の原因となるため歯科的管理が重要です。
肥満傾向と成長パターン
RSTSでは出生時の体重・身長は正常範囲のことが多いですが、生後数か月で成長曲線が急速に低下します。一方、小児期以降は肥満傾向が見られることが多く、成人の約40%が肥満となります。RSTS専用の成長曲線を用いた評価が推奨されています。

🩺 院長コラム【RSTSの行動特性について】
RSTSのお子さんは、多くの場合「友好的で社交的な性格」を持っています。これは本症候群の特徴的な行動表現型です。人懐こく、周囲との関わりを好む傾向があります。
一方で、約41%に自閉スペクトラム症(ASD)の特性が見られ、27〜64%に不安症状が認められます。ADHD様の症状(注意力低下、多動)も多く、行動面での支援が必要なこともあります。
臨床遺伝専門医として、これらの特性を理解した上で、お子さん一人ひとりに合った支援計画を一緒に考えていきます。
4. 診断方法
【結論】 RSTSの診断は臨床的特徴に基づく診断と遺伝学的検査による確定診断の組み合わせで行います。2024年に発表された国際コンセンサスの臨床診断スコアリングシステムが有用です。ただし、遺伝学的検査を行っても約30%は変異が同定されないことに注意が必要です。
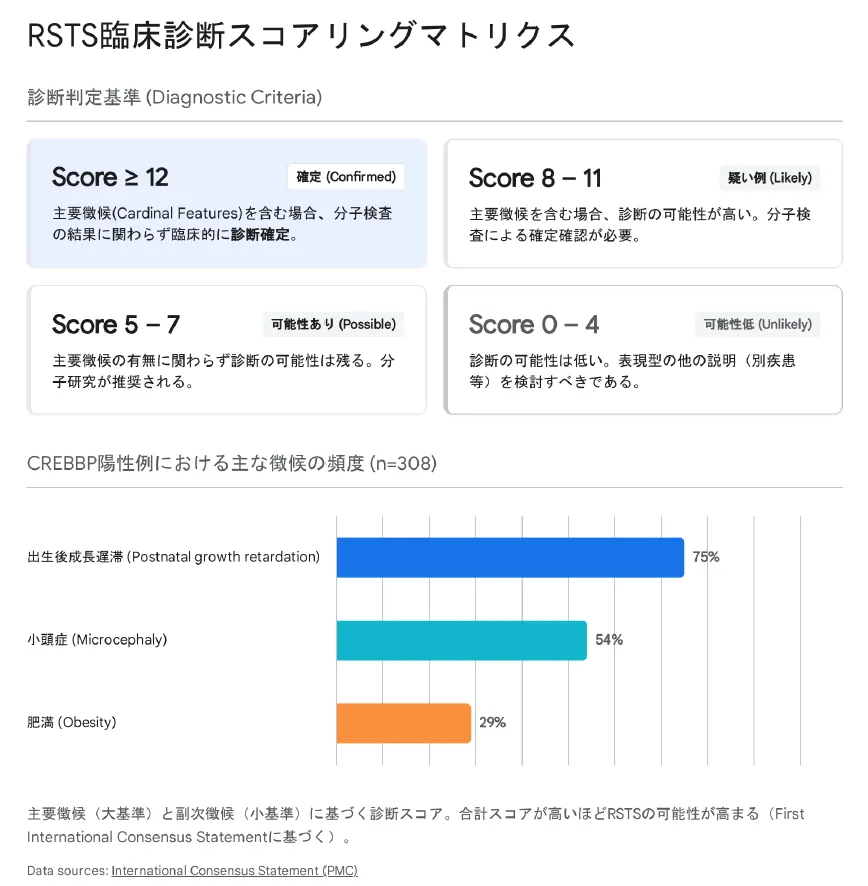
▲ RSTS臨床診断スコアリングシステム:各所見に点数をつけ、一定以上のスコアで臨床診断
臨床診断スコアリング基準(2024年国際コンセンサス)
2024年にLacombeらが発表した国際コンセンサス声明では、Cardinal Features(主要徴候)とSupportive Features(支持的徴候)に分けた点数制の臨床診断基準が提唱されました。各項目に点数を付け、合計スコアで診断の確実性を評価します。
📋 Cardinal Features(主要徴候)
1. 顔面所見(6項目中3項目以上で陽性)
- a. 弓状(アーチ型)眉毛
- b. 下外方へ傾斜した眼瞼裂
- c. 凸型の鼻背
- d. 下垂した鼻中隔(low-hanging columella)★
- e. 高口蓋
- f. 特徴的な笑顔(grimacing smile)★
配点:3項目以上陽性で3点、d・fのいずれかが陽性なら4点
2. 骨格所見
- a. 角度のついた母指・母趾(橈側偏位)★
- b. 幅広い母指
- c. 幅広い母趾
配点:b・cのいずれかが陽性で3点、aが陽性なら4点
3. 成長所見
- a. 小頭症
- b. 出生後成長遅滞
配点:aまたはbが陽性で2点
4. 発達所見
- 発達遅滞/知的障害
配点:陽性で2点
Cardinal Score判定:上記4群のうち2群以上が陽性、かつ骨格または顔面の少なくとも一方が陽性であれば「Cardinal Score陽性」
📋 Supportive Features(支持的徴候)
- a. 母体の妊娠高血圧腎症(特にEP300変異で高頻度)★
- b. ケロイド形成★
- c. 多毛症
配点:cのみ陽性で1点、aまたはbが陽性なら3点
★印:RSTSに高い特異度を持つ所見
総合スコアによる診断判定
Score ≥12
確定(Confirmed)
Cardinal Score陽性の場合、分子検査の結果に関わらず臨床的に診断確定
Score 8–11
疑い例(Likely)
Cardinal Score陽性の場合、診断の可能性が高い。分子検査による確認が必要
Score 5–7
可能性あり(Possible)
Cardinal Score陰性でも診断の可能性は残る。分子検査が推奨される
Score 0–4
可能性低(Unlikely)
診断の可能性は低い。他の疾患を検討すべき
💡 診断スコアの活用例
例:幅広い母指(3点)+下垂鼻中隔を含む顔面所見(4点)+小頭症(2点)+発達遅滞(2点)+多毛(1点)=合計12点(Cardinal Score陽性)→ 臨床的にRSTS確定となります。このような場合、遺伝学的検査で変異が見つからなくても、RSTSとして管理を行います。
診断の流れ
-
①
臨床的疑い:特徴的顔貌、幅広い母指・母趾、発達遅滞などから疑う
-
②
臨床診断スコアリング:2024年国際コンセンサス基準で評価
-
③
遺伝学的検査:CREBBP/EP300のシークエンス解析+欠失/重複解析
-
④
確定診断:病的変異の同定、または臨床診断基準を満たす
遺伝学的検査
| 検査方法 | 検出対象 | 検出率 |
|---|---|---|
| シークエンス解析 | 点変異、小欠失/挿入 | CREBBP: 50〜60%、EP300: 8〜10% |
| 欠失/重複解析(MLPA等) | 大きな欠失/重複 | 追加で5〜10% |
| 染色体マイクロアレイ(CMA) | 大きな欠失(16p13.3等) | 数% |
⚠️ 遺伝学的検査の限界
現在の検査技術をもってしても、臨床的にRSTSと診断される患者の約30%では原因変異が同定されません。これは技術的限界、モザイク、未知の原因遺伝子の存在などが理由として考えられます。変異が見つからなくてもRSTSを否定できるわけではありません。
5. 治療と長期管理
【結論】 RSTSには根本的な治療法は存在せず、症状に応じた対症療法・早期療育・継続的サーベイランスが中心となります。特に麻酔リスクとケロイド形成傾向は重要な注意点です。
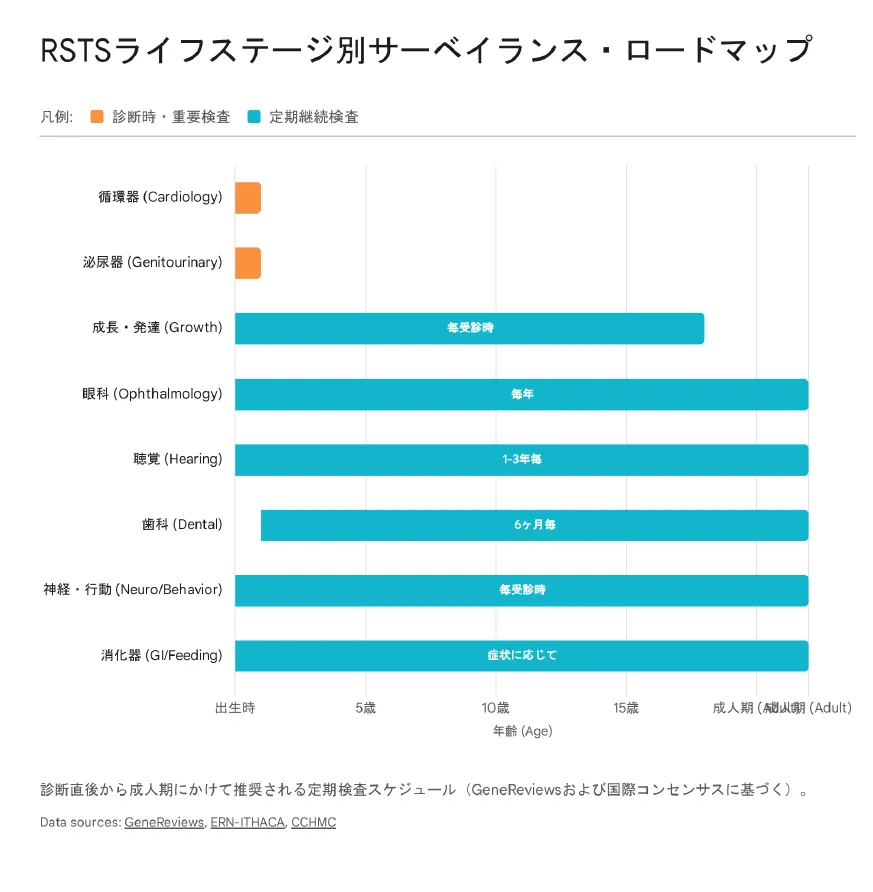
▲ RSTSライフステージ別サーベイランス・ロードマップ:各年齢で推奨される検査・評価の一覧
麻酔に関する重要な注意点
🚨 麻酔リスク:RSTS患者は麻酔ハイリスク群
RSTSの患者さんに麻酔を行う際は、以下の点に特別な注意が必要です:
1. 気道確保困難:小顎症、高口蓋、喉頭軟化症により挿管困難のリスクが高い
2. 不整脈リスク:サクシニルコリン、アトロピンの使用は避ける
3. 推奨:ロクロニウム+スガマデクスの使用が安全
手術が必要な場合は、RSTSの経験がある麻酔科医のいる施設での実施が望ましいです。
ケロイド形成傾向
RSTS患者はケロイドを形成しやすい傾向があります。このため、不必要な侵襲的処置は避けるべきであり、手術が必要な場合もケロイド予防策を講じる必要があります。小さな外傷でもケロイドになることがあるため、注意が必要です。
ライフステージ別のサーベイランス
| 領域 | 検査・評価 | 頻度 |
|---|---|---|
| 循環器 | 心エコー検査 | 診断時、その後は所見に応じて |
| 泌尿生殖器 | 腎エコー、停留精巣評価 | 診断時 |
| 成長 | 身長・体重・頭囲(RSTS専用曲線) | 1歳まで頻回、その後は定期的 |
| 眼科 | 眼科検診 | 年1回 |
| 聴覚 | 聴力検査 | 年1回(1〜3歳は特に重要) |
| 歯科 | 歯科検診 | 6か月ごと(1歳から) |
| 神経行動 | 発達評価、行動評価 | 継続的 |
| 消化器 | GERD・便秘の評価 | 継続的 |
腫瘍サーベイランス
RSTS患者では毛母腫(pilomatrixoma)の発生頻度が高く、また髄膜腫のリスクも上昇しています。ただし、40歳未満での定期的な腫瘍スクリーニングは現時点では推奨されていません。皮膚の腫瘤に気づいた場合は速やかに受診してください。
治療研究の現状
🔬 治療研究の進展
RSTSの病態がHAT活性低下によるヒストンアセチル化異常であることから、HDAC阻害薬(ヒストン脱アセチル化酵素阻害薬)による治療研究が進んでいます。
動物モデルでは、バルプロ酸やトリコスタチンAなどのHDAC阻害薬で記憶障害の改善が報告されています。しかし、ヒトでの臨床試験ではバルプロ酸による有意な認知機能改善は確認されておらず、現時点で根本治療は確立されていません。
6. 遺伝カウンセリング
【結論】 RSTSは常染色体優性(顕性)遺伝ですが、ほとんどが新生突然変異であるため、健康な両親から発症することがほとんどです。患者本人が子どもを持つ場合は50%の確率で遺伝します。
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常(新生突然変異) | 1%未満(生殖細胞モザイクの可能性) |
| 片親がモザイク保因者 | モザイク率に依存 |
| 患者本人が子を持つ場合 | 50% |
-
①
遺伝形式の説明:常染色体優性(顕性)だが、ほとんどが新生突然変異
-
②
再発リスク:両親が健康なら1%未満、生殖細胞モザイクに注意
-
③
予後の説明:90%以上が成人まで生存、知的障害の程度は様々
-
④
出生前診断の選択肢:家族内に変異が判明している場合、出生前診断は可能
🩺 院長コラム【RSTSの遺伝カウンセリングで伝えたいこと】
RSTSと診断されたお子さんの親御さんから、「なぜうちの子が?」「次の子も同じですか?」というご質問をよくいただきます。
まず知っていただきたいのは、RSTSの大多数は新生突然変異(de novo)であり、親御さんの責任ではないということです。誰にでも起こりうる「偶発的な出来事」なのです。
次のお子さんへの再発リスクは通常1%未満ですが、稀に生殖細胞モザイク(親の卵子や精子の一部だけに変異がある状態)の可能性があります。ご希望があれば、次の妊娠時に出生前診断を行うことも可能です。
臨床遺伝専門医として、ご家族の不安に寄り添い、正確な情報提供と意思決定支援を行っています。
7. 出生前診断について
【結論】 家族内でRSTSの原因変異が判明している場合、羊水検査・絨毛検査による出生前診断が可能です。ただし、ほとんどのRSTSは新生突然変異で発症するため、事前に家族歴がないケースでの偶発的発見は稀です。
出生前検査の選択肢
| 検査 | 対象 | 備考 |
|---|---|---|
| 羊水検査・絨毛検査 | 家族内変異が判明している場合 | 確定診断可能 |
| NIPT | 一般的なスクリーニング | RSTSの直接検出は困難 |
| 胎児超音波 | 形態異常の評価 | 母指の異常、心疾患などが見つかることも |
⚠️ NIPTでのRSTS検出について
現在のNIPT(新型出生前診断)は主に染色体の数的異常をスクリーニングする検査であり、CREBBP/EP300の点変異や小さな欠失を直接検出することはできません。16p13.3の大きな欠失が見つかる可能性はありますが、RSTSの大部分を占める点変異は検出対象外です。
なお、ミネルバクリニックのCOATE法を用いたNIPTでは12種類の微小欠失症候群をスクリーニングできますが、RSTS(16p13.3/22q13.2)は対象に含まれていません。
予後と長期転帰
RSTSの予後は一般的に良好であり、90%以上が成人まで生存します。知的障害はありますが、適切な支援により社会生活を送ることが可能です。成人期には就労支援や生活支援を受けながら、グループホームなどで生活している方も多くいらっしゃいます。
🏥 ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医が遺伝カウンセリングから確定検査まで一貫してサポート。
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能です。
互助会(8,000円)により、確定検査費用も全額カバーされます。
よくある質問(FAQ)
参考文献
- [1] Stevens CA. Rubinstein-Taybi Syndrome. 2002 Aug 30 [Updated 2019 Aug 22]. In: Adam MP, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. [GeneReviews]
- [2] Hennekam RC. Rubinstein-Taybi syndrome. Eur J Hum Genet. 2006;14(9):981-985. [PubMed]
- [3] Milani D, Manzoni FM, Pezzani L, et al. Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Ital J Pediatr. 2015;41:4. [PubMed]
- [4] Fergelot P, Van Belzen M, Van Gils J, et al. Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused by EP300 mutations. Am J Med Genet A. 2016;170(12):3069-3082. [PubMed]
- [5] Van Gils J, Magdinier F, Fergelot P, Lacombe D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes (Basel). 2021;12(7):968. [PubMed]
- [6] Petrij F, Giles RH, Dauwerse HG, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376(6538):348-351. [PubMed]
- [7] Roelfsema JH, White SJ, Ariyürek Y, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76(4):572-580. [PubMed]
- [8] Wincent J, Luthman A, van Belzen M, et al. CREBBP and EP300 mutational spectrum and clinical presentations in a cohort of Swedish patients with Rubinstein-Taybi syndrome. Mol Genet Genomic Med. 2016;4(1):39-45. [PubMed]
- [9] OMIM #180849 – Rubinstein-Taybi Syndrome 1. [OMIM]
- [10] OMIM #613684 – Rubinstein-Taybi Syndrome 2. [OMIM]
関連記事







