目次

ペルオキシソームアシルCoAオキシダーゼ欠損症(ACOX1欠損症)は、体内の超長鎖脂肪酸(VLCFA)を分解するために不可欠な酵素が先天的に欠損することで、脳・肝臓・眼・耳などの多臓器に深刻なダメージが生じる極めてまれな遺伝性代謝疾患です。世界で数十例しか報告されていない超希少疾患でありながら、別名「偽新生児副腎白質ジストロフィー(pseudo-NALD)」とも呼ばれ、Zellweger症候群スペクトラムやX連鎖副腎白質ジストロフィー(X-ALD)との正確な鑑別が、適切な医療につながる重要な鍵を握っています。
Q. ペルオキシソームアシルCoAオキシダーゼ欠損症とはどのような疾患ですか?まず結論だけ知りたいです
A. ACOX1遺伝子の両方に病的変異が生じることで、ペルオキシソーム内の脂肪酸分解酵素(ACOX1)が欠損し、超長鎖脂肪酸(VLCFA)が脳・肝臓などに蓄積する常染色体劣性遺伝の超希少疾患です。新生児期から重篤な筋緊張低下・けいれん・肝腫大・網膜障害が現れ、現時点で根治療法は存在せず、多くの場合、幼少期に重篤な経過をたどります。
- ➤疾患の定義 → OMIM #264470、Orphanet ORPHA:2971、別名「偽新生児副腎白質ジストロフィー(pseudo-NALD)」
- ➤分子メカニズム → ACOX1酵素欠損→VLCFA蓄積→酸化ストレス・脱髄・神経炎症カスケード
- ➤主な症状 → 新生児期の筋緊張低下・けいれん・肝腫大・網膜ジストロフィー・難聴・脳白質病変
- ➤鑑別診断 → Zellweger症候群スペクトラム・X-ALD・ミッチェル症候群との違いを詳解
- ➤診断・管理 → 血漿VLCFA検査・ACOX1遺伝子解析と多職種連携による総合ケアの実際
1. ペルオキシソームアシルCoAオキシダーゼ欠損症とは:疾患の定義と歴史的背景
ペルオキシソームアシルCoAオキシダーゼ欠損症(ACOX1欠損症)は、ACOX1遺伝子の両対立遺伝子に病的変異が生じることで、ペルオキシソームにおける超長鎖脂肪酸(VLCFA)のβ酸化が障害される先天性代謝異常症です。国際疾患データベースOMIMには「#264470」、Orphanetには「ORPHA:2971」として登録されており、常染色体劣性遺伝形式をとります。
💡 用語解説:ペルオキシソームとは
ペルオキシソームは、細胞の中にある小さな袋状の細胞小器官(オルガネラ)です。特に炭素数22以上の超長鎖脂肪酸の分解・胆汁酸合成・プラズマローゲン(特殊な脂質)の生成など、生命維持に欠かせない代謝反応を担います。また、代謝産物として生じる有害な過酸化水素(H₂O₂)を無毒化する働きも持っています。ACOX1欠損症ではペルオキシソームの形態自体は正常ですが、内部の重要な酵素が欠損することで機能が損なわれます。これがZellweger症候群との最大の違いです。
この疾患の別名である「偽新生児副腎白質ジストロフィー(pseudo-neonatal adrenoleukodystrophy:pseudo-NALD)」という名称には重要な歴史的背景があります。かつて「新生児副腎白質ジストロフィー(NALD)」という病名が、新生児期から発症する重篤な神経疾患群に使われていましたが、その多くは現在Zellweger症候群スペクトラム(PEX遺伝子変異によるペルオキシソーム形成障害)に再分類されています。ACOX1欠損症はこれらと極めてよく似た症状を示すものの、ペルオキシソームの構造は正常に存在し、あくまで単一酵素(ACOX1)のみの欠損である点が本質的に異なります。「偽(pseudo)」という接頭辞は、この「構造正常・機能欠損」という病態を的確に反映したものです。
本疾患が独立した疾患実体として初めて記載されたのは1988年、Poll-Théらによる報告です。「ペルオキシソームが正常に存在するにもかかわらず、acyl-CoA oxidase活性が選択的に欠損する」という新たな疾患カテゴリーを確立し、その後のペルオキシソーム病の分類体系に大きな影響を与えました。世界的に極めてまれな疾患であり、文献上の報告例は数十例にとどまります。
💡 用語解説:常染色体劣性遺伝(じょうせんしょくたいれっせいいでん)
「常染色体」とは、性染色体(X・Y)以外の22対の染色体のことです。「劣性(潜性)」とは、両方の染色体に変異があって初めて症状が現れる遺伝形式を意味します。両親がそれぞれ1つずつ変異を持つ「保因者」である場合、通常は発症しません。ACOX1欠損症では父親と母親の両方から変異遺伝子を受け継いだ場合(確率25%)に発症します。保因者になる確率は50%、両変異ともに受け継がない確率は25%です。
2. 原因遺伝子ACOX1と分子病態メカニズム
ACOX1欠損症の病態を理解するうえで核心となるのは、「なぜ特定の脂肪酸が蓄積するのか」「なぜそれが脳や神経を傷つけるのか」というメカニズムです。この理解が、正確な診断と適切な管理の根拠となります。
ACOX1遺伝子:ペルオキシソームβ酸化の「入口の扉」を担う律速酵素
💡 用語解説:ペルオキシソームβ酸化とは
β酸化とは、脂肪酸を段階的に切り崩してエネルギーを取り出す化学反応のことです。ミトコンドリア(細胞のエネルギー工場)でも行われますが、ペルオキシソームでは特に炭素数22以上の超長鎖脂肪酸(VLCFA)の分解を専門的に担います。この「ペルオキシソームβ酸化」の第一ステップ(最初の反応)を触媒するのがACOX1酵素です。ACOX1が欠損すると、VLCFAはここで行き詰まり、分解されないまま体内に蓄積し始めます。
ACOX1遺伝子(Acyl-CoA Oxidase 1)は第17番染色体長腕(17q25.1)に位置し、ペルオキシソームβ酸化の律速酵素(反応速度全体を決定する最初のステップの酵素)をコードしています。この酵素が正常に機能することで、食事由来や体内で合成された超長鎖脂肪酸は適切にペルオキシソーム内で処理されます。
ACOX1欠損症を引き起こす変異は、ミスセンス変異(アミノ酸の置き換わり)・ナンセンス変異・フレームシフト変異・スプライスサイト変異など多様なタイプが報告されています。いずれも機能喪失型(loss-of-function)変異であり、ACOX1酵素活性の著明な低下または完全消失をもたらします。これは、後述するミッチェル症候群を引き起こす「機能獲得型(gain-of-function)」変異とは本質的に逆のメカニズムです。
VLCFA蓄積から神経炎症カスケードへ:多段階の毒性メカニズム
💡 用語解説:超長鎖脂肪酸(VLCFA)とは
脂肪酸は炭素原子が鎖状につながった分子です。炭素数が22以上のものを「超長鎖脂肪酸(Very Long Chain Fatty Acids:VLCFA)」といいます。代表的なものはヘキサコサン酸(C26:0)です。VLCFAは通常、ペルオキシソームβ酸化によって速やかに分解されますが、ACOX1が欠損すると血漿・脳・肝臓・副腎などに異常蓄積し、強い毒性を発揮します。血液検査でVLCFAを測定することが、最初のスクリーニング検査として活用されます。
ACOX1酵素が欠損すると、VLCFAがペルオキシソームβ酸化の第一ステップで滞り、全身の組織に蓄積します。VLCFAは以下の複合的なメカニズムで神経系・肝臓を傷害します。
- ①ミエリン鞘への直接毒性:VLCFAは神経線維を包む「ミエリン鞘」の脂質成分に過剰に取り込まれ、ミエリンの物理的安定性を崩して脱髄(ミエリンの崩壊)を促進します
- ②酸化ストレスの亢進:ACOX1の基質(VLCFAのアシルCoA)が酸化される際に過酸化水素(H₂O₂)が産生されます。過剰になると活性酸素種(ROS)が細胞膜・タンパク質・DNAを酸化傷害します
- ③神経炎症カスケードの誘発:VLCFAはミクログリア(脳の免疫細胞)を過剰活性化させ、TNF-αなどの炎症性サイトカインを産生させます。この炎症カスケードが進行性の白質破壊をもたらします
- ④ミトコンドリア機能障害:VLCFAはミトコンドリアの膜電位を乱し、ATP合成(エネルギー産生)を阻害します。エネルギー需要の高い神経細胞はこの障害に特に脆弱です
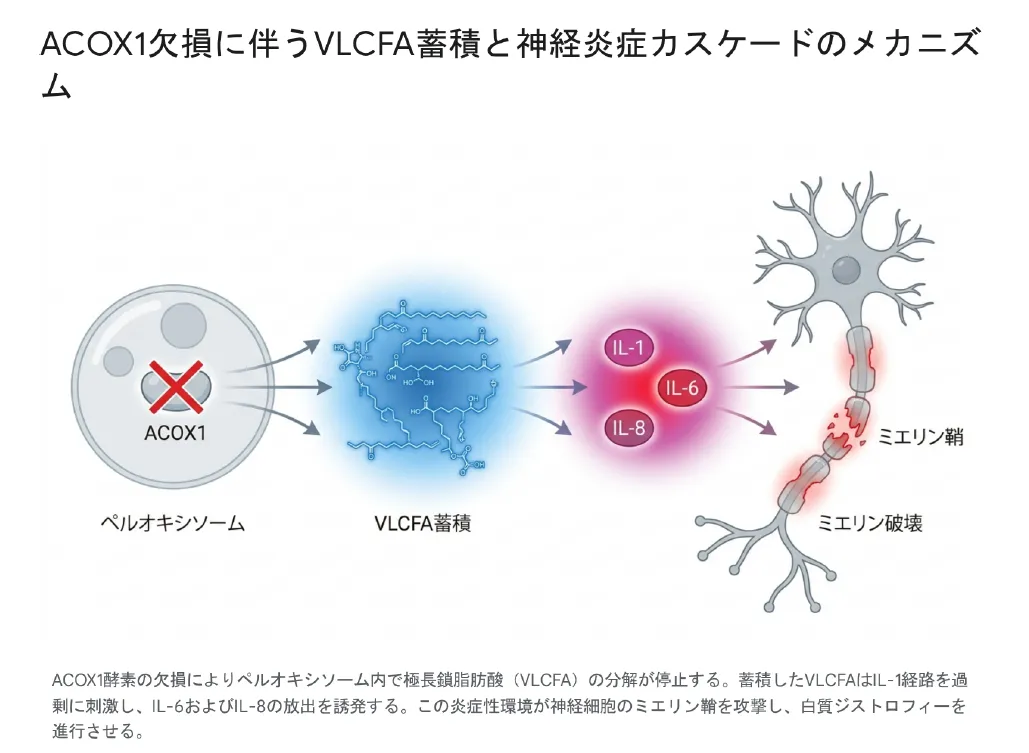
ACOX1欠損によりペルオキシソームβ酸化が障害されるとVLCFAが蓄積し、酸化ストレス・ミクログリア過活性化・炎症性サイトカイン産生を通じた神経炎症カスケードが脳白質を進行性に破壊するメカニズム。
ペルオキシソームβ酸化経路におけるACOX1の位置づけ
(VLCFA)
⚠️ ACOX1が欠損すると→VLCFAが行き詰まり体内に蓄積→脳・神経・肝臓などに毒性が現れる
ACOX1機能喪失(欠損症)とACOX1機能獲得(ミッチェル症候群):同じ遺伝子の正反対の変異が全く異なる疾患を生む
ACOX1遺伝子には、機能喪失とは逆の「機能獲得型(Gain-of-Function)」変異による全く異なる疾患が近年発見されています——それがミッチェル症候群です。ACOX1欠損症(機能喪失型・常染色体劣性)では生まれつき酵素が著しく低下するのに対し、ミッチェル症候群ではACOX1が「過剰に活性化」し続けることで、H₂O₂の過剰産生と持続的な神経炎症がきたされます。同じ遺伝子でも変異の方向性によって、発症年齢・遺伝形式・VLCFA値・臨床像がすべて異なります。
3. 主な症状と臨床経過
ACOX1欠損症の症状は新生児期〜乳児期早期にかけて出現し、神経・感覚器・肝臓など多臓器に及びます。報告例のほぼすべてで進行性の神経学的退行が見られ、予後は一般に重篤です。
主要症状の出現頻度(報告例の集計より概算)
ほぼ全例
~95%
~90%
~85%
~80%
~75%
~65%
一部例
※症例数が少なく文献によって頻度に幅があるため概算。
🧠 神経系
- 全般的な筋緊張低下(フロッピーインファント):新生児期から
- 難治性けいれん・てんかん:抗てんかん薬に抵抗性を示すことが多い
- 脳白質ジストロフィー:MRIで進行性の脱髄像・白質信号異常
- 精神運動発達の停滞・退行:一時的な発達獲得の後、機能喪失が進行
- 末梢神経障害(脱髄性ニューロパチー)
👁️ 感覚器・眼
- 網膜ジストロフィー:視力障害・夜盲・視野狭窄
- 眼振(ニスタグムス):眼球の不随意運動
- 感音性難聴:蝸牛・聴神経の障害
- 眼底色素異常(色素性網膜炎様変化)
🫀 消化器・内分泌
- 肝腫大・肝機能障害:VLCFAの肝臓内蓄積に起因
- 消化管の吸収障害・哺乳不良:新生児期
- 副腎機能不全:一部の症例で認められる
- 成長障害
📋 身体所見・その他
- 顔面の軽微な形態異常:高い額・扁平な顔・眼の離れ(眼距離開離)など
- 骨点状石灰化(一部):X線で骨端部に石灰化像
- 腎嚢胞(一部)
- Zellweger症候群と異なり重篤な骨格変形・眼窩異常は少ない
💡 用語解説:脱髄(だつずい)・脳白質ジストロフィー
神経線維を保護・絶縁する「ミエリン鞘(さや)」が傷つき、崩壊する現象を脱髄といいます。ミエリンは電気信号の高速伝達に必須であり、これが失われると、筋肉の動き・感覚・認知機能などすべての神経機能が低下します。白質ジストロフィーとは、脳の「白質(神経線維が密集する部分)」のミエリンが進行性に壊れていく疾患群の総称です。MRIのT2強調画像で白く見える異常信号として捉えることができます。
臨床経過の特徴:一時的な「ハネムーン期」と進行性退行
ACOX1欠損症の臨床経過において特筆すべきは、一部の患者で乳児期に一時的な発達獲得(寝返り・座位・初語など)が見られる点です。この安定期は数か月から数年続く場合があり、家族にとって「改善した」と感じられることもあります。しかしその後、けいれんの頻度増加・運動機能の喪失・視力・聴力の低下といった進行性の退行が始まります。この「ハネムーン期」の存在が、Zellweger症候群(ほぼ発達獲得なく重篤)との臨床的差異の一つとなっています。

4. 鑑別診断:似ている疾患との見分け方
ACOX1欠損症は症状が他のペルオキシソーム病・白質ジストロフィーと重複するため、正確な鑑別診断には生化学的検査と遺伝子解析の組み合わせが不可欠です。
Zellweger症候群スペクトラムとの鑑別
共通点:VLCFA上昇・筋緊張低下・肝腫大・けいれん・神経発達障害・感覚器障害
鑑別ポイント:Zellweger症候群ではペルオキシソーム構造自体が欠如(PEX遺伝子変異)し、より重篤な顔面奇形・骨格変形・腎嚢胞・肝臓病変を示し、多くは生後1年以内に死亡。ACOX1欠損症はペルオキシソーム構造正常・単一酵素欠損で、プラズマローゲンやフィタン酸は正常範囲のことが多い。
X連鎖副腎白質ジストロフィー(X-ALD)との鑑別
共通点:VLCFA上昇・脳白質病変・副腎機能不全
鑑別ポイント:X-ALDはABCD1遺伝子のX連鎖性変異によるもので、男児に重篤(女性は保因者)。発症年齢・脳MRIの病変分布パターン・副腎機能不全の有無・遺伝形式が異なる。ABCD1遺伝子検査が決定的。
ミッチェル症候群との鑑別
共通点:神経炎症・白質病変・けいれん・ACOX1遺伝子が関与
鑑別ポイント:ACOX1欠損症は機能喪失型・常染色体劣性・VLCFA高値、ミッチェル症候群は機能獲得型・常染色体顕性(またはde novo)・VLCFAは正常〜軽度上昇が多い。発症年齢・遺伝形式・ACOX1酵素活性の違いが重要。ACOX1遺伝子の変異の種類と機能的評価が決め手。
その他の先天性代謝異常との鑑別
グルコーストランスポーター1(GLUT1)欠損症、ミトコンドリア呼吸鎖疾患、スフィンゴ脂質代謝異常症など
鑑別ポイント:血漿・赤血球VLCFAの上昇(特にC26:0、C26:0/C22:0比)はペルオキシソーム病に特異的であり、他の代謝疾患との最初の鑑別に有用。
💡 用語解説:プラズマローゲン・フィタン酸
プラズマローゲンは、ペルオキシソームで合成される特殊な脂質で、赤血球や神経細胞膜に多く含まれます。ペルオキシソームの形成障害(Zellweger症候群スペクトラム)では著明に低下します。一方、ACOX1欠損症ではプラズマローゲンは正常に維持されます。フィタン酸は食事由来の分岐脂肪酸で、ペルオキシソームで代謝されます。これも単純なACOX1欠損症では通常正常範囲です。これらの生化学的指標の組み合わせが鑑別の助けになります。
5. 診断の進め方:生化学的検査と遺伝子解析
ACOX1欠損症の診断は、①生化学的スクリーニング → ②ペルオキシソーム機能の詳細評価 → ③ACOX1遺伝子解析という段階的なアプローチで進めます。
第1ステップ:血漿VLCFA測定(最初のスクリーニング)
ACOX1欠損症の診断において最初かつ最も重要なスクリーニング検査は、血漿超長鎖脂肪酸(VLCFA)の測定です。ヘキサコサン酸(C26:0)の絶対値上昇と、C26:0/C22:0比の上昇が特徴的な所見です。新生児・乳児期の原因不明の筋緊張低下・けいれん・肝腫大を認める症例では、早期にVLCFA測定を検討すべきです。
🔬 ACOX1欠損症を疑うべき臨床的レッドフラッグ(組み合わせが重要)
- ➤新生児〜乳児期の原因不明の高度筋緊張低下
- ➤難治性けいれん(抗てんかん薬に抵抗性)
- ➤肝腫大・黄疸・肝酵素上昇(新生児期)
- ➤眼科所見:眼振・網膜色素変性様変化
- ➤MRIで脳白質異常(T2高信号・後頭部〜頭頂部優位のことが多い)
- ➤軽微な顔面形態異常(Zellweger類似だが軽度)
第2ステップ:ペルオキシソーム機能の詳細評価
VLCFAが上昇していた場合、次に以下の検査でペルオキシソーム病の種類を絞ります。プラズマローゲン(赤血球)・フィタン酸・ピペコリン酸・胆汁酸中間体を測定し、ACOX1欠損症ではこれらが正常〜軽度異常(Zellweger症候群では著明に異常)であることを確認します。必要に応じて皮膚線維芽細胞によるACOX1酵素活性の直接測定が行われ、確定的な機能的根拠を得ます。
第3ステップ:ACOX1遺伝子解析による確定診断
💡 用語解説:全エクソーム解析(WES)・遺伝子パネル検査
全エクソーム解析(Whole Exome Sequencing:WES)は、ゲノム中のタンパク質をコードするすべてのエクソン領域を一度に解析する手法です。複数の代謝疾患・白質ジストロフィーを一括して鑑別できるため、診断確定に要する期間を大幅に短縮できます。遺伝子パネル検査は、特定の疾患群(ペルオキシソーム病・白質ジストロフィーなど)に関連する複数の遺伝子を同時に解析します。どちらも患者本人と両親の3名(トリオ)で行うと変異の病的意義の解釈がより正確になります。
ACOX1遺伝子の両アレル(対立遺伝子)に病的変異が同定されることで確定診断となります。変異の解釈にはACMGの最新ガイドラインを用い、機能的アッセイ(酵素活性・タンパク質発現)の結果を組み合わせて病原性を判定します。特に発見された変異がVUS(意義不明の変異)と分類されるケースでは、酵素活性の直接測定が診断確定の鍵を握ります。
なお、ACOX1欠損症と類似したVLCFA上昇・神経症状を呈するACADVL欠損症(超長鎖アシルCoA脱水素酵素欠損症)・MFP2欠損症(ペルオキシソーム多機能タンパク質2型欠損症)なども鑑別に挙がり、これらを含む遺伝子パネル検査の活用が推奨されます。
6. 治療と長期管理
ACOX1欠損症に対して現時点では根治的な治療法は存在しません。治療の中心は、各症状への対症療法と合併症予防のための集学的管理です。神経・眼科・消化器・代謝の各専門科が連携した多職種チームによるケアが不可欠です。
けいれん・てんかん管理
各種抗てんかん薬(バルプロ酸・レベチラセタム・クロナゼパムなど)が使用されますが、薬剤抵抗性を示すことが多く、複数薬剤の組み合わせによる発作コントロールが中心となります。発作の性状・脳波評価を定期的に行い、用量を調整します。
食事療法(Lorenzo’s Oil)
X-ALDで有名な「Lorenzo’s Oil(ローレンツォオイル:オレイン酸・エルカ酸の混合物)」は血漿VLCFAを低下させますが、ACOX1欠損症での有効性に関するエビデンスは限られています。個々の症例で有益性・有害性を検討のうえ導入を判断します。
リハビリテーション・感覚支援
理学療法・作業療法・言語聴覚療法による発達支援を継続します。視覚障害に対するロービジョンケア、感音性難聴に対する補聴器・人工内耳の検討、嚥下障害へのアプローチも重要な管理の柱です。
定期的なモニタリング
血漿VLCFAの定期測定、肝臓超音波・肝酵素、脳MRI(白質病変の進行評価)、眼底検査・視覚誘発電位、聴性脳幹反応(ABR)、副腎機能評価(血清コルチゾール・ACTH)を定期的に行い、病勢と臓器障害の進行を把握します。
今後の治療展望:遺伝子治療・酵素補充療法の研究動向
ACOX1欠損症の根治に向けた研究は、動物モデルを用いたAAVベクターを用いた遺伝子補充療法や、mRNA療法の開発が国際的に進められています。また、神経炎症を標的とした抗炎症治療(ACOX1欠損症の病態に共通するミクログリア過活性化を抑制するアプローチ)の可能性も検討されています。世界的に報告例が少ないため、国際的な患者レジストリへの登録が治療開発を加速させる基盤として重要です。
7. 遺伝カウンセリングの意義
ACOX1欠損症の確定診断後、ご家族への丁寧な遺伝カウンセリングが必要です。遺伝カウンセリングで扱われる主な内容は以下の通りです。
- ➤再発リスクの説明:常染色体劣性遺伝のため、両親がともに保因者である場合、次子の罹患リスクは25%です。罹患しない子どもの2/3は保因者となります。
- ➤保因者診断:両親・同胞(兄弟姉妹)の保因者検査を実施し、家族内の遺伝リスクを整理します。保因者は通常無症状ですが、将来の子どもへの影響を理解するうえで重要な情報です。
- ➤出生前診断の選択肢:次子を望む保因者カップルには、絨毛検査・羊水検査による出生前遺伝子診断が選択肢として存在します。既知の病的変異が両親で同定されている場合は確実な診断が可能です。
- ➤心理的サポートと情報提供:疾患の超希少性から国内情報が限られています。Zellweger症候群・X-ALDなどペルオキシソーム病全般の患者家族会の情報提供、国際的な研究への参加についても情報を共有します。
- ➤長期的な医療連携:希少疾患センターや大学病院の代謝内科・神経内科との長期的な連携体制を構築し、治療研究の進展に応じた情報を随時提供します。
8. よくある誤解
誤解①「Zellweger症候群と同じ病気」
Zellweger症候群はペルオキシソーム自体の形成障害(PEX遺伝子変異)であり、ACOX1欠損症はペルオキシソーム構造は正常で単一酵素のみが欠損する別カテゴリーの疾患です。臨床的には似ていますが生化学的・遺伝的に明確に異なります。
誤解②「VLCFAが高ければX-ALD」
VLCFAの上昇はペルオキシソーム病全般に見られます。X-ALD・ACOX1欠損症・Zellweger症候群など複数の疾患でVLCFAは上昇します。遺伝子検査・酵素活性・プラズマローゲンなど複合的な評価が必要です。
誤解③「ミッチェル症候群と同じ」
同じACOX1遺伝子に関わりますが、機能喪失(欠損症)か機能獲得(ミッチェル症候群)かで病態・遺伝形式・VLCFAの程度・治療の方向性がすべて異なります。変異の種類と機能的評価が不可欠です。
誤解④「親が健康だから遺伝ではない」
常染色体劣性遺伝のため、保因者である両親は通常まったく無症状です。「両親が健康だから遺伝病ではないはず」という思い込みが診断を遅らせる典型的なパターンです。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少遺伝性代謝疾患の診断・遺伝カウンセリングについて
ACOX1欠損症をはじめとするペルオキシソーム病・希少代謝疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
参考文献
- [1] Poll-Thé BT, et al. A new peroxisomal disorder with enlarged peroxisomes and a specific deficiency of acyl-CoA oxidase (pseudo-neonatal adrenoleukodystrophy). Am J Hum Genet. 1988;42(3):422-434. [PubMed]
- [2] Ferdinandusse S, et al. ACOX1 mutations in acyl-CoA oxidase deficiency (pseudo-neonatal adrenoleukodystrophy). Hum Mutat. 2007;28(2):207. [PubMed]
- [3] Wanders RJA, Waterham HR. Biochemistry of Mammalian Peroxisomes Revisited. Annu Rev Biochem. 2006;75:295-332. [PubMed]
- [4] Orphanet. Acyl-CoA oxidase deficiency. ORPHA:2971. [Orphanet]
- [5] OMIM #264470. PEROXISOMAL ACYL-CoA OXIDASE DEFICIENCY. Johns Hopkins University. [OMIM]
- [6] Morais P, et al. Mitchell syndrome: a new ACOX1 gain-of-function variant disorder. Brain. 2021;144(12):e101. [PubMed]
- [7] Waterham HR, et al. Biochemical aspects of peroxisomal biogenesis disorders. Biochimie. 1994;76(3-4):139-144. [PubMed]
- [8] Moser HW, et al. Adrenoleukodystrophy and other peroxisomal disorders that affect the nervous system, including new observations on L-carnitine administration. Ann Neurol. 1984;16(5):628-641. [PubMed]
- [9] NCBI Gene. ACOX1 acyl-CoA oxidase 1 [Homo sapiens]. Gene ID: 51. [NCBI Gene]






