目次
CACNA1C遺伝子は、脳や心臓など多くの組織で重要な役割を果たしている電位依存性カルシウムチャネルのサブユニットをコードする遺伝子です。この遺伝子の変異は、ティモシー症候群、ブルガダ症候群、QT延長症候群、知的障害を伴う神経発達障害など、様々な疾患を引き起こすことが知られています。
本記事では、CACNA1C遺伝子の基本情報から関連する疾患、遺伝子検査の重要性まで詳しく解説します。知的障害や自閉症スペクトラム障害、不整脈などの症状でお悩みの方々にとって、遺伝的背景を理解することは適切な医療を受けるための第一歩となります。
CACNA1C遺伝子とは
CACNA1C遺伝子は、正式名称を「Calcium Channel, Voltage-Dependent, L Type, Alpha-1C Subunit」といい、12番染色体の短腕(12p13.33)に位置しています。この遺伝子は、L型電位依存性カルシウムチャネル(CaV1.2)のポアを形成するα-1Cサブユニットをコードしています。
CaV1.2チャネルは、心臓、肺、脳、平滑筋など多くの組織で発現しており、以下のような重要な機能を担っています:
- カルシウムシグナル伝達
- 細胞および神経の興奮性の調節
- 筋肉の収縮
- 遺伝子発現の制御
特に脳の発達過程において、CACNA1C遺伝子は神経細胞の分化や移動、シナプス形成など重要な役割を果たしています。このため、この遺伝子の変異は知的障害や自閉症など様々な神経発達障害の原因となることがあります。
CACNA1C遺伝子の構造
CACNA1C遺伝子は非常に複雑な構造を持っており、約150キロベースにわたって広がっています。この遺伝子は44の不変エクソンと6つの選択的エクソンから構成されており、多様なスプライシングバリアントを生成することができます。
CaV1.2チャネルタンパク質は、4つの繰り返しドメイン(I〜IV)から構成され、各ドメインには少なくとも6つの膜貫通領域(S1〜S6)が含まれています。これらのドメインは可変長のリンカーで接続されています。中でも重要なのは以下の構造的特徴です:
- S4セグメント:電位センサーとして機能
- S5-S6ループ:イオン選択性フィルターを形成
- N末端領域:チャネル制御に関与するNSCaTE(N端空間的カルシウム変換要素)を含む
- C末端領域:カルモジュリン結合サイトを含み、カルシウム依存性不活性化に重要
特に重要なのは、CACNA1C遺伝子の選択的スプライシングによる多様性です。例えば、エクソン8とエクソン8Aは相互排他的に選択され、組織特異的な機能を持つチャネルバリアントを生成します。心臓と脳では主にエクソン8を含むバリアントが発現しており(全体の約80%)、これらの組織における正常な機能に重要です。
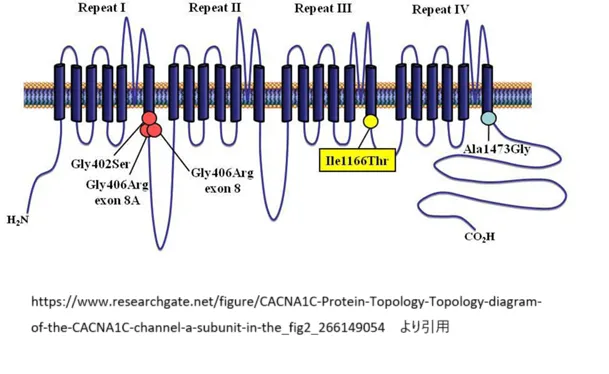
https://www.researchgate.net/figure/CACNA1C-Protein-Topology-Topology-diagram-of-the-CACNA1C-channel-a-subunit-in-the_fig2_266149054
より引用
重要情報
米国医学遺伝学会(ACMG)は、発達遅延、知的障害、先天異常に対して、ゲノムまたはエクソーム解析を第一選択の検査とすることを推奨しています。早期の遺伝子検査は適切な治療介入や家族計画に役立ちます。
CACNA1C遺伝子の機能
CACNA1C遺伝子がコードするCaV1.2チャネルは、細胞膜の脱分極に応じてカルシウムイオンの流入を制御する重要なタンパク質です。その主な機能は以下の通りです:
心臓における機能
心筋細胞において、CaV1.2チャネルは活動電位のプラトー相を維持し、興奮-収縮連関に重要な役割を果たしています。具体的には:
- 心筋細胞の収縮を開始するための細胞内カルシウム濃度上昇
- リアノジン受容体を介したカルシウム誘発性カルシウム放出の制御
- 心臓の自動能と刺激伝導系の調節
脳における機能
神経細胞において、CaV1.2チャネルは以下のような重要な機能を持っています:
- シナプス伝達の調節
- 神経可塑性の制御
- CREB(cAMP応答配列結合タンパク質)やMEF2などの転写因子の活性化
- 神経細胞の生存と分化の制御
特に興味深いのは、CaV1.2チャネルのC末端領域が切断されると、「カルシウムチャネル関連転写制御因子(CCAT)」として核内に移行し、直接遺伝子発現を制御することができる点です。これにより、電気信号と遺伝子発現の間に直接的なリンクが形成されます。
血管平滑筋における機能
血管平滑筋細胞では、CaV1.2チャネルは以下の機能を持っています:
- 筋収縮と血管緊張度の調節
- 血圧の維持
- 組織血流の制御
CACNA1C遺伝子関連疾患
ティモシー症候群
ティモシー症候群は、CACNA1C遺伝子の特定のエクソンにおける変異(主にG406R変異)によって引き起こされる多臓器障害を特徴とする症候群です。主な症状には以下が含まれます:
- 致命的な不整脈
- 手足の合指(指の間の皮膚のつながり)
- 先天性心疾患
- 免疫不全
- 間欠的な低血糖
- 認知機能障害
- 自閉症
この症候群では、カルシウムチャネルの不活性化が障害されることで、細胞内へのカルシウム流入が過剰になり、様々な組織でカルシウム過負荷を引き起こします。特に心臓では、心筋細胞の再分極遅延を引き起こし、不整脈のリスクを高めます。
ブルガダ症候群3型
CACNA1C遺伝子の変異は、ブルガダ症候群3型の原因となることがあります。この症候群は心電図上の特徴的な所見と共に、突然死のリスクを高める不整脈症候群です。特にQT間隔が短縮している場合があります。
QT延長症候群8型
QT延長症候群8型は、CACNA1C遺伝子の変異により心電図上のQT間隔が延長する疾患です。失神発作や突然死のリスクがあります。
低緊張、言語発達遅延、骨格異常を伴う神経発達障害(NEDHLSS)
この疾患は、CACNA1C遺伝子の非切断型変異(主にミスセンス変異)によって引き起こされる神経発達障害です。主な特徴として以下が挙げられます:
- 筋緊張低下(低緊張)
- 言語発達の遅れ
- 骨格の異常
- てんかん発作(症例によって異なる)
- 知的障害
この疾患では、心臓の症状がないか、あっても軽度であることが多く、神経発達の問題が前面に出ています。
注意事項
米国遺伝カウンセラー協会(NSGC)は、原因不明のてんかんを有するすべての患者に対して、ゲノムまたはエクソーム解析を第一選択検査とすることを推奨しています。この推奨は米国てんかん学会(AES)にも支持されています。
これらのバリアントは、チャネルの機能に様々な影響を与えます。例えば、ティモシー症候群に関連するバリアントは主にチャネルの不活性化を阻害し(機能獲得型変異)、神経発達障害に関連するバリアントは様々な機序で作用します。
ヒント
RNA統合シークエンス解析(RNA-ISE)検査は、通常のエクソーム解析やゲノム解析で診断がつかなかった患者さんの約20%で疾患原因を特定できる可能性があります。特に複雑なケースでは、より詳細な解析が役立つことがあります。
CACNA1C遺伝子検査の重要性
CACNA1C遺伝子の検査は、以下のような場合に特に重要です:
- 原因不明の知的障害がある
- 自閉症スペクトラム障害と心臓症状の両方がある
- 家族歴に突然死や不整脈がある
- 発達遅滞と共に特徴的な身体的特徴(合指症など)がある
- 原因不明のてんかん発作がある
遺伝子検査によってCACNA1C遺伝子の変異が確認されることで、以下のようなメリットがあります:
- 正確な診断の確立
- 適切な医療管理計画の策定
- 将来的なリスクの予測
- 家族への遺伝カウンセリングの提供
- 特定の治療法の検討(例:カルシウムチャネル遮断薬など)
特に心臓症状を伴う場合は、適切な診断と管理によって突然死のリスクを軽減できる可能性があります。
専門家のアドバイス
遺伝子検査の結果解釈は複雑であるため、検査前後に適切な遺伝カウンセリングを受けることが重要です。ミネルバクリニックでは臨床遺伝専門医が常駐しており、専門的なアドバイスを提供しています。
ミネルバクリニックにおける遺伝子検査
ミネルバクリニックでは、CACNA1C遺伝子を含む様々な知的障害・自閉症関連遺伝子の検査を提供しています。以下のような検査オプションがあります:
知的障害遺伝子検査パネル
知的障害の原因となる主要な遺伝子を一度に検査することができます。CACNA1C遺伝子も含まれており、効率的に原因遺伝子を特定することが可能です。
自閉症遺伝子検査パネル
自閉症スペクトラム障害の遺伝的背景を調べるための検査です。CACNA1C遺伝子を含む多くの自閉症関連遺伝子を一度に解析します。
発達障害・自閉症・知的障害染色体シーケンス解析
より広範囲な染色体異常を調べることができ、大きな欠失や重複などの構造的変異も検出できます。
RNA統合シークエンス解析(RNA-ISE)検査
通常のDNA検査で診断がつかなかった場合に有効な高度な検査法です。RNAレベルでの異常を検出することで、通常の検査では見逃される変異を特定できることがあります。
当クリニックでは、臨床遺伝専門医が検査の選択から結果の解釈、その後の対応まで一貫してサポートしています。遺伝子検査は保険適用されません。自費診療になります。
まとめ:CACNA1C遺伝子と遺伝カウンセリングの重要性
CACNA1C遺伝子は、神経発達や心臓機能に重要な役割を果たしており、その変異は複数の重要な疾患を引き起こします。特に知的障害や自閉症スペクトラム障害、心臓不整脈などの症状がある場合は、この遺伝子の検査が診断の助けになる可能性があります。
遺伝子検査を受ける際には、結果の解釈や今後の対応について専門家からの適切なアドバイスを受けることが重要です。ミネルバクリニックでは、最新の遺伝子検査技術と専門的な知識を提供し、患者さんとそのご家族をサポートしています。
遺伝子検査に関するご質問や不安がある方は、ぜひ一度ミネルバクリニックの遺伝カウンセリングをご利用ください。一人ひとりの状況に合わせた適切なアドバイスを提供いたします。
遺伝子検査で不明な発達の課題を解明する
お子様の発達の遅れや知的障害の原因がわからずお悩みの方は、遺伝子検査が答えを見つける手助けとなる可能性があります。CACNG2遺伝子を含む多くの遺伝子が知的障害に関与していることがわかっています。
ミネルバクリニックでは、最新の遺伝子検査技術を用いて、原因となる遺伝子変異を特定するサポートを行っています。臨床遺伝専門医による丁寧な説明と、検査結果に基づいた適切な医療・療育につなげるお手伝いをします。
参考文献
- Bozarth, X., et al. (2018). Novel de novo CACNA1C variant in a patient with Timothy syndrome, cardiac defects, and severe developmental delay. American Journal of Medical Genetics Part A.
- Splawski, I., et al. (2004). CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell.
- Antzelevitch, C., et al. (2007). Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation.
- Rodan, L. H., et al. (2021). CACNA1C-related disorders: Genotype-phenotype correlations of calcium channelopathies. American Journal of Medical Genetics Part A.
- Panagiotakos, G., et al. (2019). Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. eLife.







