目次
自閉症スペクトラム障害(発達障害)遺伝子パネル検査参照論文1
自閉症スペクトラム障害児における染色体>マイクロアレイ解析と全エクソームシークエンシングの分子診断率
https://jamanetwork.com/journals/jama/fullarticle/2432161
自閉症スペクトラム障害児における染色体マイクロアレイ解析と全エクソームシークエンシングの分子診断結果
抄録
重要:
自閉症スペクトラム障害(ASD)患者に対する分子診断を提供するためのゲノムワイド検査の使用には、さらなる研究が必要である。
目的:
発達期小児臨床に典型的なサンプルにおけるこれらの検査の分子診断率を決定するために、ASDの小児の不均一なグループにおいて染色体マイクロアレイ分析(CMA)および全エクソーム配列決定(WES)を実施すること。
デザイン、設定、および参加者 サンプルは、主要な先天異常および軽度の身体的異常の存在に基づいて形態スコアを定義するために詳細な評価を受けた、連続して確認されたASDの非血縁小児258人から構成された。カナダのニューファンドランドとラブラドールで2008~2013年に子供を募集した。発端者を、形態学的重症度の増加する3群:本態性、不明瞭性、および複雑性(0~3、4~5、および≧6のスコア)に層別化した。
曝露すべての確率はCMAを行い、WESは95の確率親トリオに対して行った。
主なアウトカムと測定 3つの表現型群に層別化した集団ベースのASDサンプルにおけるCMAおよびWESの全体的な分子診断率。
結果:
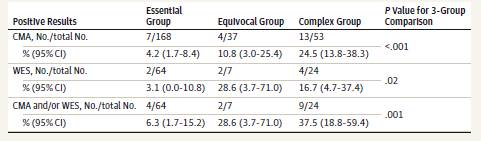
発端者258人のうち、24人(9.3%、95% CI、6.1%~13.5%)がCMAから、95人のうち8人(8.4%、95% CI、3.7%~15.9%)がWESから分子診断を受けた。結果は形態群間で統計学的に異なっていた。CMAでは、検査結果陽性の小児の割合は、本態性群で168例中7例(4.2%、95% CI、1.7%~8.4%)、不明瞭群で37例中4例(10.8%、95% CI、3.0%~25.4%)、複合群で53例中13例(24.5%、95% CI、13.8%~38.3%)であった(P < 0.001)。WESについては、割合はそれぞれ64人中2人(3.1%、95% CI、0.0%-10.8%)、7人中2人(28.6%、95% CI、3.7%-71.0%)、24人中4人(16.7%、95% CI、4.7%-37.4%)であった(3群比較、P = .02)。CMAおよびWES検査の両方を受けた小児の間で、同定可能な遺伝的病因を有する推定割合は15.8%(95% CI、9.1%-24.7%;小児15/95人)であった。これには、両検査から分子診断を受けた2人の小児が含まれた。総合分子診断率は、本態性群(小児64例中4例)で6.3%(95% CI、1.7%~15.2%)、不明瞭群(小児7例中2例)で28.6%(95% CI、3.7%~71.0%)、複合群(小児24例中9例;3群比較、P=0.001)で37.5%(95% CI、18.8%~59.4%)であった。複合診断率は本態性群と比較すると複合群で有意に高かった(対比較、P = .002)。
結論および関連性:ASDの小児の不均一なサンプルの中で、CMAおよびWESの分子診断率は同等であり、複合分子診断率は、本態性カテゴリーの小児と比較して、より複雑な形態学的表現型を有する小児で高かった。追加の集団で再現された場合、これらの所見はASDに罹患した小児に対する分子診断検査の適切な選択に情報を与える可能性がある。
序文:
自閉症スペクトラム障害(ASD)は有病率が増加する神経発達状態の一群を表す。1 臨床像および転帰はASD.2において実質的に異なる。中核的な自閉症的特徴のばらつきに加えて、多くの罹患者は医学的、認知的、および精神的健康の共存症を有する。 3 ASDの小児は、集団の5%未満に存在する形態学的偏位として定義される軽微な身体的異常の過剰を有するというエビデンスもある。 4,5
ASDの広範な表現型スペクトラムは基礎にある遺伝的病因にも反映されており、これは同定可能な単一遺伝子症候群から大きな染色体不均衡まで様々である。 6染色体マイクロアレイ解析(CMA)は、収率が7.0%8~9.0%のASD7の個人に対する第一段階遺伝子検査として推奨される。 ASD患者の研究コホートに関する9 Whole-exome sequencing (WES)は、これらの努力にもかかわらず、ASDの病因における配列レベルの新規変異を強調している。OEにもかかわらず、不均一なASD試料におけるWESの分子診断率は未定義であり、これらの同じ患者に関するCMAデータは臨床診療に情報を提供するのに役立つ可能性がある。
ASDの表現型の複雑さは依然として課題であり、異なる表現型尺度を用いた層別化は、ASD患者を遺伝子検査から利益を得られる可能性が高いサブタイプに分類するのに役立つ可能性がある。 12,13Milesらの13は、臨床形態分類を用いて、多発性の軽微な身体異常の存在に基づいて、ASDの小児の20%が「複雑」と定義されることを示した。ASDの小児の形態異常の層別化がゲノムワイド検査の分子診断とどのように関連するかについての情報も限られている。最近、ASDを有する個人のサブセットは、破壊的なde novoおよび稀なコピー数変異体(CNV)および配列レベル突然変異を有する可能性が高いことが示されている。 11,14,15
本稿では、臨床的表現型により層別化した、発達期小児科診療所で見られる小児に典型的なASD小児の集団ベースサンプルにおけるCMAおよびWESの分子診断収率について報告する。
方法
本研究のサンプルは、カナダのニューファンドランドおよびラブラドールの小児を対象とし、ASDの集学的チーム評価を実施している同州の発達期小児科クリニックの両方から2008年から2013年まで連続的に紹介された。それぞれの評価は、発達小児科医が主導し、応用行動分析療法の資金を子供に受けさせることが求められた。各子供は、精神障害の診断と統計マニュアル(第4版、テキスト改訂)からの基準に基づいてASD診断を受け、これは自閉症診断観察スケジュールと自閉症診断面接改訂評価によって確認された。すべての小児の両親または保護者は書面によるインフォームドコンセントを行い、本研究はMemorial Universityのヒト研究倫理当局の承認を得た。本稿およびその補遺で特定の症例に割り当てられた研究番号は、参加者からの特定情報とは関係がなく、コード化されているとみなすべきである。
臨床評価と形態分類
小児の家族歴および医療記録(放射線学および脳波報告を含む)をレビューした。未実施の場合は、Wechsler Preschool and Primary Scale of Intelligence IIIまたはWechsler Abbreviated Scale of Intelligenceを用いた脳磁気共鳴画像法およびIQ検査を提案した。児の標準的な身体診察に基づいて、先天異常の他のスクリーニングを整理した。形態学的検査は、形態異常学者(B.A.F.)が実施し、身長、体重、頭囲の測定;顔面、手、足の測定;および軽微な身体的異常の存在の記録(補遺のeMethods)を含めた。 16後者は、正常集団の5%未満に存在するわずかな形態学的偏位である。 4例としては、単一の手掌のしわや低位の耳がある。
Milesらによると、13では、それぞれの小児に軽微な身体的異常スコア(発生学的に無関係な軽微な身体的異常ごとに1点、および両親には存在しなかった平均値から2 SDを超える測定異常ごとに1点)を割り当てた(補遺のeMethods)。スコアを用いて、発端者を身体診察のみに基づいて、まず3つの形態学的グループのうちの1つに分類した:本態性(小身体異常スコア0~3)、不明瞭(小身体異常スコア4または5)、または複雑(小身体異常スコア≧6)。 13また、各子供には主要な先天異常スコア(各構造的脳異常または脳以外の主要な先天異常について2点)と総形態スコア(軽微な身体異常+主要な先天異常スコア)を割り当てた。全形態スコアを用いて、各子供は再度、同じカットオフ値をもつ本態性、不確定、または複雑に分類された(補遺のeFigure 1)。総形態スコア分類はCMAおよびWES分析と相関した。形態学的カテゴリーは遺伝子検査に先立って割り当てられた。
臨床検査・形態分類
家族には、発達小児科医(C.V.、V.C.、S.L.、T.D.)が主導する集学的チームが自閉症スペクトラム障害(ASD)の診断後に本研究への参加を提案した。
症候学的特徴または既知または疑わしい症候群に基づいて除外は行わなかった。
全ての患者の両親/保護者は、文書によるインフォームドコンセントを提供した。
本研究に募集された全ての発端者(年齢範囲1.5~16歳)を臨床遺伝学者が検査し、先天異常および軽微な身体異常の有無について詳細な評価を受けた。
発端者の出生前および周産期の病歴、病歴および家族歴を、放射線学およびEEG報告を含めてレビューした。
先天異常の他のスクリーニングは、発端者の標準的な身体診察に基づいて配列され、これには心血管検査(例えば、心室中隔欠損に一致する雑音を伴う発端者の心エコー図)が含まれた。
経験豊富な形態学者(B.A.F)が全症例について詳細な形態学的検査を行った。
頭蓋、顔面、眼、鼻、好中球、口、中咽頭、耳、頸部、胸部、骨盤、背部、四肢、生殖器、皮膚、毛および歯を含む、各身体部分の相対的な大きさおよび/または形状について評価した。1
可能な限り、両親とも同じ方法で検査した。
臨床写真を撮影した。
発端者のASDは、孤立性または社会的相互作用技能の低下と関連して、表現的言語技能の喪失が認められた場合に、退行性発症であると分類された。
すべての臨床データを関係データベースに入力した。
各測定値を年齢に換算し、利用可能であれば性特異的なセンチル値とした。1
身体診察の異常はロンドンを用いて標準化した
Dysmorphology Databaseはコード化し(n=683)、次の3群に割り付けられた:1)集団の5%未満に起こる軽微な身体的異常(例:単一の手掌のしわ、過剰に折りたたまれた耳のらせん);2)平均値からの2標準偏差を超える測定異常(例:眼の多眼症、大きな耳);および3)しばしば家族性で容易に測定できない集団の4%以上に起こる記述的形質(深く設定された眼、広い額)(Milesらの記載のように)。
各発端者は、発生学的に無関係なMPA、測定異常、または親には存在しない記述的異常の各々に対して1点を与えることにより、「Minor Physical Anomaly」(MPA)スコアを割り当てられた(例えば、前頭隆起を伴う頭囲>+2標準偏差(SD)に対して1点が割り当てられるであろう)。
両親が検査に利用できない場合には、写真の見直しを試みた。
MPAスコアを用いて、各発端者を「本態性」(MPAスコア0~3)、「不明瞭」(MPAスコア4または5)、または「複雑」(MPAスコア6以上)の3群の1つに分類した。
カルテの見直しと放射線画像の結果に基づいて、各発端者をMajorに割り当てた
脳外の発生学的に無関係な先天性欠損ごとに2点の先天異常(MCA)スコア。
脳MRIを受けた患者にはBrain Anomalyスコアを与え、発生学的に無関係な脳の構造的異常ごとに2点とした。
また、各発端者は、形態異常検査と先天異常の有無(総形態スコア= MPAスコア+ MCAスコア+/脳奇形スコア)の組み合わせを用いて分類した。
この分類はMPAのみのスコアと同様に行われた。
分子遺伝学
DNA抽出および/または樹立リンパ芽球様細胞系のための全血を、各発端者、利用可能な両親、および同胞から採取した。各発端者は脆弱X症候群の検査を受けた。全ての女児はMECP2配列決定を受けており、平均値より3 SD以上の頭囲を有する全ての小児はPTEN配列決定を受けていた。臨床的に何らかの症候群が疑われる場合は、関連する標的配列決定をオーダーした。 17
分子遺伝学的解析は補遺のeFigure 1に要約されている。染色体解析は、クリニカルマイクロアレイ、前述の高分解能(>1Mプローブ)リサーチマイクロアレイ、18-21、またはその両方を用いて実施した(補遺のeMethods参照)。
コピー数変異体はAmerican College of Medical Genetics and Genomicsガイドラインに従って分類した。 22 病原性または変異型-有意性不明-病原性と分類されたコピー数の変化については、定量的ポリメラーゼ連鎖反応法および蛍光in situハイブリダイゼーション法を用いて確認および親の検査を完了した。 両親からのDNA試料が入手可能であった23100の発端者を、それらのCNV状態を知らずに無作為に選択した。発端者と両親は、Ion AmpliSeq Exome Kit (Life Technologies)によるエキソン増幅後、Ion Protonシステムを用いてWESを受けた。品質管理後、さらに95トリオを分析した(補遺のeTable 1)。母集団データベース(国立心肺血液研究所エキソーム24および1000ゲノム25)で頻度が1%未満の稀な変異体を優先し、推定ASD関連変異体を検索した。さらなる優先順位付けは、以前にASDまたは他の神経発達障害21,26に関与していた遺伝子の一覧表および徒手治癒(補遺のeMethod)に基づいて行った。その後、American College of Medical Genetics and Genomicsのガイドラインを用いて変異体を分類した。 病原性または意義不明の病原性と分類されたOBOnly変異体-可能性の高い病原性-を分子診断率の算出に含めた。
主なアウトカム
測定した主なアウトカムは、ASD発端者における表現型の差、CMAおよびWESからの分子診断の収率、および形態学的グループ間の分子診断収率の差であった。
統計解析
表現型特性と分子診断の収率の差の比較は、事後対比較によるFisher直接確率検定を用いて行った。対検定のP値は、Holm調整を用いて3回の同時比較で補正した。分子診断の割合の95%信頼区間は二項分布に基づいて算出した。形態学的群間の診断時年齢およびIQの差は、1元配置分散分析(ANOVA)を用いて検定し、続いて対比較のためのT統計およびP値のHolm補正を行った。群間の検出されたde novo変異体の有病率の95%信頼区間および差の計算は、Poisson検定を用いて行った。α = .05の有意水準を用い、すべての検定は両側とした。3群間の比較はいずれも推定検出力が0.9を超えていた。推定された統計的検出力の計算については、補遺のeMethodsに記載されている。すべての統計解析はRソフトウェアversion 3.2.0を用いて実施した。
結果
試験サンプルには、書面によるインフォームドコンセントが得られた連続的に紹介された小児258人が含まれた(表1)。紹介されたすべての家族で、参加を拒否したのは10%未満であった。小児27例(10.5%)、143例(55.4%)、88例(34.1%)が、それぞれアスペルガー症候群、自閉性障害、広汎性発達障害に分類されたが、他に特定されなかった。CMAおよびWESの前に、発端者258人中12人(4.7%)もまた、標的配列決定を伴うまたは伴わない身体診察に基づいて、臨床的に異なる症候群と診断された。これらの症候群のうち1例を除く全てがASD関連(258例中11例、4.3%)に分類され、その内訳は、PTEN (OMIM 601728)変異を有する女児1例と、ASD感受性にも寄与したと考えられるホモ接合性WNT1(OMIM 164820)変異28による骨形成不全症の女児1例であった(補遺のeTable 2)。 29
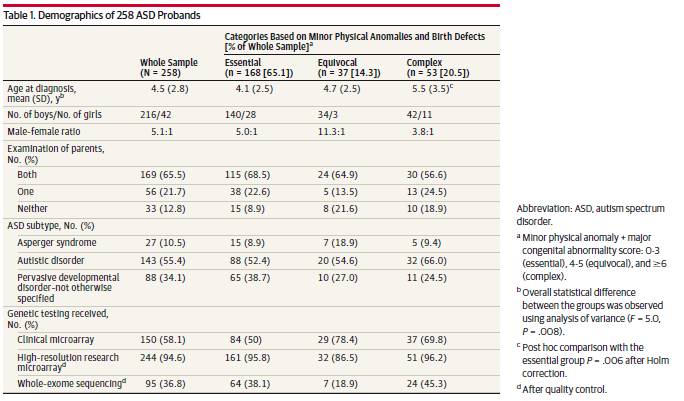
マイナーな身体的異常スコアを用いたカテゴリー的アプローチ(すなわち、形態異常検査のみに基づく)に基づき、発端者の179人(69.4%)は必須と分類され、51人(19.8%)は不明瞭な形態異常を有し、28人(10.9%)の発端者は複雑であった(表2)。小児127例(49.2%)にIQ検査を行い、109点について得点を得た。IQ値は3群間で同等であった(Table 2)。女子の得点の分布は男子よりも二相性の表現が多かったが(eFigure 2B)、女子と男子では統計的な差は認められなかった(補遺のeFigure 2A)。軽微な身体的異常スコアで層別化した3つの形態学的群は、小児の44.6%(115/258)に基づく脳波異常の結果(P = .03)、サンプルの63.6%(164/258)に基づく脳の構造的異常(P < .001)、および脳以外の主要な先天異常(P < .001)の小児数において統計学的に異なっていた(フィッシャー直接確率検定を用いた3群比較)(表2)が、これまでの所見と一致している。 13,30The 対群比較の結果、複合群の小児はこれらの測定値が最も異なり、複合群では充実していた(表2)。そこで、事後設定において、最終的な形態学的カテゴリーには、脳の構造異常や脳以外の主要な先天異常の有無を含めた総形態スコアを用いた(Table 1)。カテゴリー化のための総スコアを用いて、複合体として分類された発端者は10.9%から20.5%(53/258)に増加した。ASDサブタイプまたは平均IQスコアにカテゴリー群間で統計学的な差は認められなかったが、診断時の平均年齢は群間で異なり(1元配置分散分析、P = .008)、複合群で診断時の平均年齢が遅かった(表1、補遺のeFigure 2)。
CMA診断
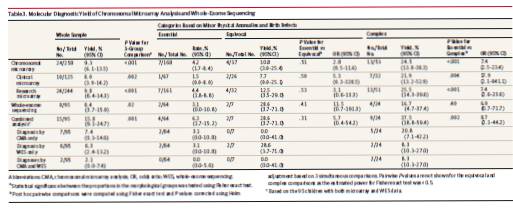
本研究の過程で、258人の小児全員が何らかの形態のマイクロアレイ検査を受けた。小児150例(58.1%)に臨床マイクロアレイ検査を実施し、そのうち125例はオリゴヌクレオチドアレイを用いて実施した(追加の小児25例については、低分解能細菌人工染色体マイクロアレイに基づく比較ゲノムハイブリダイゼーションを実施した)。発端者の94.6%(244/258)に対して高分解能(>1Mプローブ)研究マイクロアレイ遺伝子型タイピングを実施した(表1)。発端者24例における計24の分子診断が、発端者258例の全CMAから同定された(9.3%、95% CI、6.1%-13.5%) (補遺の表3およびeTable 3)。分子診断の結果は、研究用マイクロアレイサンプルで9.8%(95% CI、6.4%~14.3%、24/244)、臨床用マイクロアレイサンプルで8.0%(95% CI、3.9%~14.2%、10/125)であった(表3)。臨床マイクロアレイ検査を受けた125人のうち、114人は高解像度研究マイクロアレイデータも持っていた。研究マイクロアレイ解析により、これらの小児にさらに3つの臨床的に重要なCNVが認められた。定義された形態学的カテゴリー内の分子診断の数を分析し、3つの形態学的グループにわたるCMAの診断率の差を実証した(Fisher直接確率検定、P < .001)(表3)。
複合ASD群は、本態性群(7/168、4.2%、95% CI、1.7%~8.4%; Holm調整P < .001)と比較して、病原性CNVが有意に多かった(13/53、24.5%、95% CI、13.8%~38.3%)。親の検査が可能であった臨床的に重要な変異体については、57.1%(12/21)が発端者におけるde novo事象であった(補遺のeTable 3);これらのde novo事象の12例中11例が複合カテゴリーの発端者で検出された。母系遺伝は23.8%(5/21)、父系遺伝は変異体の19%(4/21)であった。
WESからの分子診断
WESデータ(100トリオ)の品質管理後、95人の発端者をさらに分析した。これらについては、平均被覆深度108×が達成され、配列決定の標的となるエクソン領域の90.5%が少なくとも20×カバーされていた(補遺のeTable 1)。95人の発端者のうち、9つの突然変異を有する8人の小児がASD関連分子診断を受けた(8.4%、95% CI、3.7%-15.9%) (補遺のeTable 4);これらの小児のうち2人はすでにCMAから1分子診断を受けていた(症例3-0075-000および3-0095-000、補遺のeTable 3および4)。WESから分子診断を受けた発端者の割合は形態群間で異なり、収量は複合群で16.7%(95% CI、4.7%~37.4%、4/24)、不確定群で28.6%(95% CI、3.7%~71.0%、2/7)、本態性群で3.1%(95% CI 0.0%~10.8%、2/64; Fisher直接確率検定、P = 0.02)であった(表3)。9変異の遺伝様式は、常染色体優性(de novo変異3例を含む)6例(66.7%)、常染色体劣性2例(22.2%)、X連鎖劣性1例(11.1%)であった。
ASD dysmorphologyによる配列レベルのde novo変異体
合計96のde novo変異体が同定され、WESの小児95例中55例で確認された(小児あたり0~5のde novo変異、補遺のeTable 5)。コード配列に影響を及ぼすde novo変異体の数は、血液由来DNAが利用可能な小児89例(補遺のeMethodsおよびeFigure 3)における軽微な身体的異常および重大な先天異常の存在に基づく総形態スコアと弱い相関を示した(r = 0.2、P = .03)。複合群では、本態性群と比較して有意な増加(Poisson検定、P = .02)が観察され(1.0イベント/小児、95% CI、0.6~1.5)、症候群小児ではde novoイベントの負荷が高いことを示した(図)(0.55イベント/小児、95% CI、0.4~0.8)。最も高い有病率は複合群の女児で検出され(小児あたり1.75件、95% CI、0.7-3.6)、これは本態性群の女児よりも有意に高かった(小児あたり0.33件、95% CI、0.07-1.0; Poisson検定、P = .01)。いずれの比較においても女子と男子の間に有意差は認められなかった。
96のde novo変異体のうち、3は分子診断につながる変異として分類された(補遺のeTable 4および5)。これらには、ASDおよび知的障害性変異体ASH1L (c.C7189T, OMIM 607999)およびWAC (c.576_585delGCAAGCAACA, OMIM 615049)における2つの機能喪失ミューテーション、およびSCN2A (OMIM 182390)におけるde novo ミスセンスミューテーションが含まれた。
遺伝配列レベルの変異体
分子診断に至った9変異のうち6変異が遺伝した(66.6%) (補遺のeTable 4)。発端者3‐111‐000では、Bardet‐Biedl症候群1(BBS1)遺伝子(OMIM 209901)にホモ接合性M390Rミスセンス変異(c.T1169G)が認められた。生後22ヵ月の登録時に、詳細不明の過成長症候群(補遺のeTable 2)の診断を受けたが、WES後の臨床的再評価後、非定型Bardet-Biedl症候群の診断が確認された。TCF12(OMIM 600480)の中の早すぎたストップコドンに至る、妊産婦に継承されたフレームシフト挿入(c.1106_1107g)は、近心症や左前頭部皮質形成不全の領域を含む、疑わしい変形をもつ確率3-0459-000で検出された。また、FGFR2(OMIM 176943)において別の推定機能損失ミューテーション(c.A1295G)を検出した。この発端者(3~211~000)はまた、円頭症、深部設定眼、および左側頭葉中隔実質内囊胞を含む不明瞭な形態異常を有した;しかし、頭蓋骨癒合症の特徴はなかった。
WESからの偶発的および医学的に行動可能な所見
発端者95例中8例(8.4%)で偶発的または医学的に実行可能な所見が報告され(補遺のeTable 6)、そのすべてが遺伝性突然変異であった。6例(6.2%)は医学的に利用可能とみなされた。すなわち、追加のベースライン臨床試験または進行中のスクリーニングにより、罹病率または死亡率に関して治療成績の改善が期待される結果であった。これら6つの結果を家族に伝えた。偶発的所見には、SDHB (OMIM 185470)およびCACNA1S (OMIM 114208)に生じた3つの突然変異が含まれ、それぞれ家族性傍神経節腫および悪性高熱症を引き起こした。
CMAとWESの複合収率
WESを受けた発端者95人中9人(9.5%)はすでにCMAから分子診断を受けており、WESの発端者はCNVの状態を知らずにランダムに選択された。このCMA陽性率は、サンプル全体(9.30%)と同程度である(表3)。そこで、この発端者のサブセットを用いて、CMAおよびWES後に同定可能な遺伝的病因を有するASD患者の割合を推定したところ、15.8%(95% CI、9.1%-24.7%、15/95)であった。2名の小児が両検査から分子診断を受け、いずれも複合群であった(表3)。異なる形態群における総合収率は、本態性で6.3%(95% CI、1.7%~15.2%、4/64)、不確定で28.6%(95% CI、3.7%~71.0%、2/7)、複合群で37.5%(95% CI、18.8%~59.4%、9/24)であった(Fisher直接確率検定、P = .001)(表3)。複合収量は本態性群と比較すると複合群で有意に高かった(対比較、P = .002)。
考察
ASDの小児の集団ベースのサンプルでは、WESの分子診断率(8.4%)はCMAからの収率(9.3%)と同等であり、現在、ASDの個人に対する第一選択の遺伝子検査として推奨されている。7,31A併用分子診断率15.8%が、両検査を受けた小児に認められた(表3)。
臨床的に重要なCNVについて観察された収率(9.3%)は、8,9我々の研究で用いられた高解像度マイクロアレイと、新たな候補遺伝子に影響を及ぼす稀な変異体のより良好な解明のために、以前の報告よりもわずかに高い。 32,33では、急性骨髄性白血病は未だ主に研究領域である。 10,11 いくつかの臨床WES研究は、追加の医学的状態を伴うより小さな自閉症サブグループに対する分子診断推定値を提供しており、これらは様々であった(0%~45.0%)。 34-36今回の研究では、診察に基づくASD発端者の形態学的層別化に関連する差異を実証した。今回のデータから、ASD小児の医学的評価は、遺伝子検査により分子診断を達成する可能性が高い集団を同定するのに役立つ可能性があることが示唆される。形態学的層別化は分子診断の収率に関連しており、分析を複雑な表現型の提示を有する個人のサブセットに限定した場合、CMAおよびWESの両方でより高かった。CMAとWESの複合診断率の分析に基づいて、著者らは、付加的な医学的および形態異常の特徴を有するASD小児の35%以上が分子診断を受けることができるかもしれないと推定する。対照的に、症候学的特徴のないASD小児のうち、われわれの研究で分子診断を受けたのはわずか6.0%であった(表3)。知的障害を併存している本態性ASDの小児を除外すると、分子診断率はさらに低くなる可能性がある。我々の分析では、CMAまたはWESのいずれかによる分子診断を受けた本態性群の小児4人中2人が知的障害を併存していた(補遺のeTable 7)。ここに示したデータは、過去10年間に発生したASDの小児に対する分子診断の指定における改善にも焦点を当てている。2005年、Milesらは13、自閉性障害の小児260例を3つの形態学的群に層別化して発表し、複雑な指定は不良な治療成績を予測するための特異度が87%であることを示した。彼らの解析では、4.2%(小児260人中11人)が臨床検査および染色体分析により同定可能な遺伝的症候群を有していた。 13我々は、2005年の結果と比較して、分子診断率(15.8%)が3.5倍以上増加していることを示している。比較を複雑な群に限定すると、診断率はマイルズらの13で23.9%(11/46)、我々の研究で37.5%であった(表3)。
ASDの小児の遺伝子検査は増加し続ける可能性が高いと思われる。親のASD遺伝子検査への関心度を調査したところ、37 80%の親は、検査によって診断が確認できなかったり、排除できなかったりしても、ASDリスクのミューテーションを確認するために検査した2年以内に兄弟を望むであろうと述べた。 遺伝子検査の結果が正である一部の子どもに対しては、ASD関連の医療状態を対象とした治療計画を提示することができる。 ODOの例としては、それぞれ1q21.1と17q12の除去シンドロームをもつ見込みのある若年層の心臓欠陥と成熟開始糖尿病のスクリーニングがあり、16p11.2の微小削除を行っている人の肥満の発生を避けるために綿密なモニタリングがある。ASDの診断時年齢は複合群で有意に高齢であることが観察され(表1)、このことは、ASDの行動徴候について、形態学的症候群が疑われるか診断された小児の監視において、医療従事者は特に慎重である必要があることを示唆している。
我々の研究はASDの遺伝的および表現型の不均一性を例証している(補遺のeTable 4、5、および7)。 OB95人の発端者の標本は、9つのASD感受性遺伝子においてWESによって見出された遺伝性およびde novo突然変異からの寄与を有し、そのうちのいくつかは、様々な発現性および浸透度を有することが知られている。そのうち4つは常染色体優性遺伝子の機能喪失型変異であった。例えば、TCF12の突然変異が頭蓋骨癒合症患者で観察されているが、軽度の学習障害から重度の自閉症に及ぶ神経発達障害が知られている。 46-48遺伝的不均一性は45の家系内にも存在する;例えば、発端者3-0027-00はASD感受性遺伝子、SLITRK5における母性遺伝性機能喪失突然変異を有し、これはASDの兄には存在しなかった。以前の研究によれば、21,49のデータは、複合群の女児の豊富さ(表1)、IQスコアのわずかに異なった分布(補遺のeFigure 2)、およびde novo変異体の高い有病率(補遺のeTable 5)から明らかなように、女児のASDは男児のASDと遺伝的に異なっている可能性があるという結論を支持している。
我々の研究の限界としては、比較的少数の症例数のほか、同意を得た家系と低下した家系の間に存在した可能性のある臨床的差異に関連する可能性のある確認バイアス(発達期の小児科診療所での診断後に研究への参加を拒否したのは10%未満)が挙げられる。脳磁気共鳴画像法を受けた小児はわずか63.5%であり、これは本態性群に有利に我々の最終的な形態学的分類を偏らせていた可能性があり、IQ検査を受けた研究サンプルはわずか49.2%(127/258)であった。この研究の主な限界は、WES解析に組み入れられた小児はわずか36.8%(95/258)であり、これが結果に対する未測定の交絡効果につながった可能性があることである。さらに、変異体の同定には技術的および解釈上の限界があり、それらは分子診断として分類された。Whole-exomeシークエンシングは、すべてのコード配列26,50について平等なカバー範囲を提供するわけではなく、構造的バリアントを検知するための敏感さと特異性を欠いている。 45 このこととCMAの分解能限界、より大きなインデル(>20塩基対)とより小さなCNV(>20キロ塩基対)の大部分を検出できないことも限界である。 1回の実験ですべてのクラスの遺伝的変異が検出される可能性があるため、全ゲノム配列決定がASDの主要な遺伝子検査になると思われる。 OB ASD関連ゲノム突然変異、特に稀な配列レベルの変異体に対する遺伝カウンセリングは、その発現性が多様であり、浸透度が不完全であるため、しばしば困難である。 17
結論
ASDの小児の不均一なサンプルの中で、CMAおよびWESの診断収率は同等であり、複合診断収率は、本態性カテゴリーの小児と比較して、より複雑な形態学的表現型を有する小児の間で高かった。追加の集団で再現された場合、これらの所見はASDに罹患した小児に対する分子診断検査の適切な選択に情報を与える可能性がある。
リファレンス
- 1
El-Fishawy P, State MW . The genetics of autism: key issues, recent findings, and clinical implications. Psychiatr Clin North Am 2010;33:83–105.
- 2
Geschwind DH . Genetics of autism spectrum disorders. Trends Cogn Sci (Regul Ed) 2011;15:409–416.
- 3
Caglayan AO . Genetic causes of syndromic and non-syndromic autism. Dev Med Child Neurol 2010;52:130–138.
- 4
Chakrabarti S, Fombonne E . Pervasive developmental disorders in preschool children. JAMA 2001;285:3093–3099.
- 5
Icasiano F, Hewson P, Machet P, Cooper C, Marshall A . Childhood autism spectrum disorder in the Barwon region: a community based study. J Paediatr Child Health 2004;40:696–701.
- 6
Lauritsen MB, Pedersen CB, Mortensen PB . Effects of familial risk factors and place of birth on the risk of autism: a nationwide register-based study. J Child Psychol Psychiatry 2005;46:963–971.
- 7
Simonoff E . Genetic counseling in autism and pervasive developmental disorders. J Autism Dev Disord 1998;28:447–456.
- 8
Constantino JN, Zhang Y, Frazier T, Abbacchi AM, Law P . Sibling recurrence and the genetic epidemiology of autism. Am J Psychiatry 2010;167:1349–1356.
- 9
Ozonoff S, Young GS, Carter A, et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics 2011;128:e488–e495.
PubMed PubMed Central Google Scholar
- 10
Butler MG, Dasouki MJ, Zhou XP, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005;42:318–321.
- 11
Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ . Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord 2007;37:738–747.
- 12
Erlandson A, Hagberg B . MECP2 abnormality phenotypes: clinicopathologic area with broad variability. J Child Neurol 2005;20:727–732.
- 13
Morrow JD, Whitman BY, Accardo PJ . Autistic disorder in Sotos syndrome: a case report. Eur J Pediatr 1990;149:567–569.
- 14
Park JP, Moeschler JB, Davies WS, Patel PI, Mohandas TK . Smith-Magenis syndrome resulting from a de novo direct insertion of proximal 17q into 17p11.2. Am J Med Genet 1998;77:23–27.
- 15
Pearl PL, Gibson KM, Acosta MT, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003;60:1413–1417.
- 16
Chudley AE, Gutierrez E, Jocelyn LJ, Chodirker BN . Outcomes of genetic evaluation in children with pervasive developmental disorder. J Dev Behav Pediatr 1998;19:321–325.
- 17
Steiner CE, Guerreiro MM, Marques-de-Faria AP . Genetic and neurological evaluation in a sample of individuals with pervasive developmental disorders. Arq Neuropsiquiatr 2003;61(2A):176–180.
- 18
Challman TD, Barbaresi WJ, Katusic SK, Weaver A . The yield of the medical evaluation of children with pervasive developmental disorders. J Autism Dev Disord 2003;33:187–192.
- 19
Kosinovsky B, Hermon S, Yoran-Hegesh R, et al. The yield of laboratory investigations in children with infantile autism. J Neural Transm 2005;112:587–596.
- 20
Battaglia A, Carey JC . Etiologic yield of autistic spectrum disorders: a prospective study. Am J Med Genet C Semin Med Genet 2006;142C:3–7.
- 21
Abdul-Rahman OA, Hudgins L . The diagnostic utility of a genetics evaluation in children with pervasive developmental disorders. Genet Med 2006;8:50–54.
- 22
Roesser J . Diagnostic yield of genetic testing in children diagnosed with autism spectrum disorders at a regional referral center. Clin Pediatr (Phila) 2011;50:834–843.
- 23
Lathe R . Fragile X and autism. Autism 2009;13:194–197.
- 24
Reddy KS . Cytogenetic abnormalities and fragile-X syndrome in autism spectrum disorder. BMC Med Genet 2005;6:3.
- 25
Shevell MI, Majnemer A, Rosenbaum P, Abrahamowicz M . Etiologic yield of autistic spectrum disorders: a prospective study. J Child Neurol 2001;16:509–512.
- 26
Weidmer-Mikhail E, Sheldon S, Ghaziuddin M . Chromosomes in autism and related pervasive developmental disorders: a cytogenetic study. J Intellect Disabil Res 1998;42(Pt 1):8–12.
- 27
Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L . Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry 2006;11:1,18–28.
- 28
Manning M, Hudgins L ; Professional Practice and Guidelines Committee. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med 2010;12:742–745.
- 29
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–764.
- 30
Coulter ME, Miller DT, Harris DJ, et al. Chromosomal microarray testing influences medical management. Genet Med 2011;13:770–776.
- 31
Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science 2007;316:445–449.
- 32
Rosenfeld JA, Ballif BC, Torchia BS, et al. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet Med 2010;12:694–702.
- 33
Schaefer GB, Starr L, Pickering D, Skar G, Dehaai K, Sanger WG . Array comparative genomic hybridization findings in a cohort referred for an autism evaluation. J Child Neurol 2010;25:1498–1503.
- 34
Shen Y, Dies KA, Holm IA, et al.; Autism Consortium Clinical Genetics/DNA Diagnostics Collaboration. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 2010;125:e727–e735.
- 35
Bremer A, Giacobini M, Eriksson M, et al. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet 2011;156:115–124.
- 36
McGrew SG, Peters BR, Crittendon JA, Veenstra-Vanderweele J . Diagnostic yield of chromosomal microarray analysis in an autism primary care practice: which guidelines to implement? J Autism Dev Disord 2012;42:1582–1591.
- 37
Betancur C . Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res 2011;1380:42–77.
- 38
Eichler EE, Zimmerman AW . A hot spot of genetic instability in autism. N Engl J Med 2008;359(16):1685–1699.
- 39
Kumar RA, KaraMohamed S, Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 2008;17:628–638.
- 40
Weiss LA, Shen Y, Korn JM, et al.; Autism Consortium. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008;358:667–675.
- 41
Walsh KM, Bracken MB . Copy number variation in the dosage-sensitive 16p11.2 interval accounts for only a small proportion of autism incidence: a systematic review and meta-analysis. Genet Med 2011;13:377–384.
- 42
Jacquemont ML, Sanlaville D, Redon R, et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet 2006;43:843–849.
- 43
International Standards for Cytogenomics Arrays Consortium. https://www.iscaconsortium.org.
- 44
UCSC Genome Bioinformatics. http://genome.ucsc.edu.
- 45
DECIPHER v5.1. http://decipher.sanger.ac.uk.
- 46
Database of Genomic Variants. http://projects.tcag.ca/variation.
- 47
Hatton DD, Sideris J, Skinner M, et al. Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A 2006;140A:1804–1813.
- 48
Watson MS, Leckman JF, Annex B, et al. Fragile X in a survey of 75 autistic males. N Engl J Med 1984;310:1462.
- 49
Cohen IL, Sudhalter V, Pfadt A, Jenkins EC, Brown WT, Vietze PM . Why are autism and the fragile-X syndrome associated? Conceptual and methodological issues. Am J Hum Genet 1991;48:195–202.
- 50
Hammer S, Dorrani N, Dragich J, Kudo S, Schanen C . The phenotypic consequences of MECP2 mutations extend beyond Rett syndrome. Ment Retard Dev Disabil Res Rev 2002;8:94–98.
- 51
Vourc’h P, Bienvenu T, Beldjord C, et al. No mutations in the coding region of the Rett syndrome gene MECP2 in 59 autistic patients. Eur J Hum Genet 2001;9:556–558.
- 52
Beyer KS, Blasi F, Bacchelli E, Klauck SM, Maestrini E, Poustka A ; International Molecular Genetic Study of Autism Consortium (IMGSAC). Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum Genet 2002;111:305–309.
- 53
Carney RM, Wolpert CM, Ravan SA, et al. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol 2003;28:205–211.
- 54
Zappella M, Meloni I, Longo I, et al. Study of MECP2 gene in Rett syndrome variants and autistic girls. Am J Med Genet B Neuropsychiatr Genet 2003;119B:102–107.
- 55
Lam CW, Yeung WL, Ko CH, et al. Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J Med Genet 2000;37:E41.
- 56
Herman GE, Henninger N, Ratliff-Schaub K, Pastore M, Fitzgerald S, McBride KL . Genetic testing in autism: how much is enough? Genet Med 2007;9:268–274.
- 57
Schaefer GB, Lutz RE . Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genet Med 2006;8:549–556.
- 58
Young DJ, Bebbington A, Anderson A, et al. The diagnosis of autism in a female: could it be Rett syndrome? Eur J Pediatr 2008;167:661–669.
- 59
Psoni S, Sofocleous C, Traeger-Synodinos J, Kitsiou-Tzeli S, Kanavakis E, Fryssira-Kanioura H . Phenotypic and genotypic variability in four males with MECP2 gene sequence aberrations including a novel deletion. Pediatr Res 2010;67:551–556.
- 60
Ramocki MB, Peters SU, Tavyev YJ, et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol 2009;66:771–782.
- 61
Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP . PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet 2001;105:521–524.
- 62
Stein MT, Elias ER, Saenz M, Pickler L, Reynolds A . Autistic spectrum disorder in a 9-year-old girl with macrocephaly. J Dev Behav Pediatr 2010;31:632–634.
- 63
Varga EA, Pastore M, Prior T, Herman GE, McBride KL . The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med 2009;11:111–117.
- 64
McBride KL, Varga EA, Pastore MT, et al. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res 2010;3:137–141.
- 65
Buxbaum JD, Cai G, Chaste P, et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 2007;144B:484–491.
- 66
Zecavati N, Spence SJ . Neurometabolic disorders and dysfunction in autism spectrum disorders. Curr Neurol Neurosci Rep 2009;9:129–136.
- 67
Baieli S, Pavone L, Meli C, Fiumara A, Coleman M . Autism and phenylketonuria. J Autism Dev Disord 2003;33:201–204.
- 68
Wraith JE, Danks DM, Rogers JG . Mild Sanfilippo syndrome: a further cause of hyperactivity and behavioural disturbance. Med J Aust 1987;147:450–451.
- 69
Haas RH . Autism and mitochondrial disease. Dev Disabil Res Rev 2010;16:144–153.
- 70
Weissman JR, Kelley RI, Bauman ML, et al. Mitochondrial disease in autism spectrum disorder patients: a cohort analysis. PLoS ONE 2008;3:e3815.
- 71
Wilder RT, Flick RP, Sprung J, et al. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology 2009;110:796–804.
- 72
Sprung J, Flick RP, Katusic SK, et al. Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clin Proc 2012;87:120–129.
- 73
Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS ONE 2009;4:e4415.
- 74
Schaefer GB, Mendelsohn NJ . Genetics evaluation for the etiologic diagnosis of autism spectrum disorders. Genet Med 2008;10:4–12.
- 75
Mefford HC, Batshaw ML, Hoffman EP . Genomics, intellectual disability, and autism. N Engl J Med 2012;366:733–743.
- 76
Schaefer GB, Mendelsohn NJ ; Professional Practice and Guidelines Committee. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders. Genet Med 2008;10:301–305.







