目次
8q21.11欠失症候群とは?
原因・症状・診断を臨床遺伝専門医が解説
Q. 8q21.11欠失症候群とはどのような病気ですか?
A. 8番染色体長腕(8q21.11)の微細欠失により発症する極めて稀な常染色体優性(顕性)遺伝性疾患です。
有病率は100万人に1人未満と推定され、知的障害・眼瞼下垂・筋緊張低下を三徴候として、ピーターズ奇形などの眼科的異常や特徴的な顔貌を呈します。
-
➤
原因 → 8番染色体長腕21.11領域のヘテロ接合性欠失(数百kb〜数Mb) -
➤
主要責任遺伝子 → ZFHX4(眼・神経発達)、HEY1(心血管・神経) -
➤
三徴候 → 知的障害・眼瞼下垂・筋緊張低下(ほぼ全例で認める) -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
頻度 → 100万人に1人未満(極めて稀少な疾患)
1. 8q21.11欠失症候群とは|基本情報
【結論】 8q21.11微細欠失症候群(8q21.11 microdeletion syndrome, OMIM #614230)は、8番染色体長腕21.11領域のヘテロ接合性欠失により発症する極めて稀な常染色体優性(顕性)遺伝性疾患です。知的障害・眼瞼下垂・筋緊張低下を三徴候とし、ピーターズ奇形などの眼科的異常を高頻度で合併します。
💡 用語解説:「ピーターズ奇形」とは?
ピーターズ奇形(Peters anomaly)は、前眼部形成不全(Anterior Segment Dysgenesis)の一種で、角膜中央部の混濁、後部角膜実質・デスメ膜・内皮の欠損、および虹彩や水晶体と角膜の癒着を特徴とする先天性眼疾患です。胎生期における前眼部の発生異常が原因であり、緑内障や視力障害を合併することがあります。8q21.11欠失症候群は、ピーターズ奇形を伴う数少ない染色体異常の一つとして認識されており、責任遺伝子ZFHX4の機能不全が眼の発生異常に関与していると考えられています。
「検査で8q21.11欠失が見つかった」「お子さんにこの症候群が疑われている」という方は、まず正確な情報を知ることが大切です。2010年代初頭に独立した症候群として確立された比較的新しい疾患概念であり、世界で数十例程度の報告しかありません。
💡 用語解説:「ハプロ不全」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。「ハプロ不全(haploinsufficiency)」とは、1コピーが欠失または機能しなくなることで、残り1コピーだけでは正常な機能を維持できない状態を指します。8q21.11欠失症候群では、ZFHX4遺伝子やHEY1遺伝子のハプロ不全が症状の原因となります。
8q21.11欠失症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 8q21.11微細欠失症候群(OMIM #614230) |
| 原因 | 8q21.11領域の微細欠失(数百kb〜数Mb) |
| 頻度 | 100万人に1人未満(極めて稀) |
| 遺伝形式 | 常染色体優性(顕性)(多くは新生突然変異) |
| 主要責任遺伝子 | ZFHX4、HEY1、PEX2 |
| 三徴候 | 知的障害・眼瞼下垂・筋緊張低下 |
⚠️ 従来のG分染法では検出できません
8q21.11の欠失は数百kb〜数Mb程度の微細な構造異常であるため、従来のG分染法による染色体核型分析では検出が困難です。このため、本症候群は染色体マイクロアレイ検査(CMA)が普及した2010年代以降に独立した症候群として認識されるようになりました。
歴史的背景と症例報告
Palomaresら(2011年)による先駆的な症例集積研究により、本症候群の表現型プロファイルが初めて体系化されました。8人の非血縁患者の欠失領域を解析し、全員に共通する最小欠失領域(約539kb)が同定されています。
💡 隣接遺伝子症候群としての理解
8q21.11欠失症候群は単一遺伝子疾患ではなく、隣接する複数の遺伝子(Contiguous gene syndrome)がそれぞれの機能不全を通じて複合的な臨床像を形成しています。欠失範囲に含まれる遺伝子の種類によって、症状の組み合わせが異なることがあります。
2. 8q21.11欠失症候群の主な症状
【結論】 本症候群の臨床像は多岐にわたりますが、顔貌の特徴、神経発達障害、眼科的異常に関しては高い浸透率が認められます。「眼瞼下垂を伴う丸い顔」という特徴的な顔貌が診断の重要な手がかりとなります。
症状は個々の患者によって重症度や合併症の組み合わせが異なりますが、以下の特徴が高頻度で認められます。
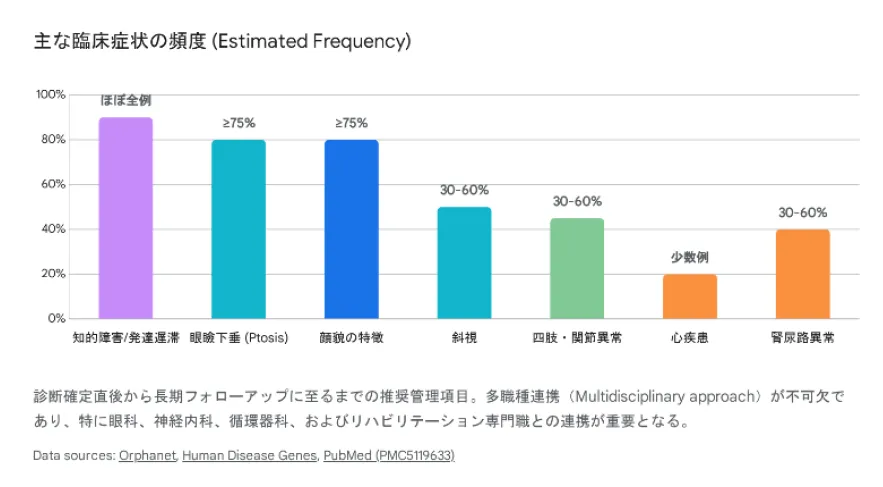
症状の出現頻度
| 症状カテゴリー | 頻度 | 詳細 |
|---|---|---|
| 知的障害・発達遅滞 | ほぼ全例 | 軽度〜中等度の知的障害、精神運動発達遅滞 |
| 眼瞼下垂 | ≥75% | 両側性が多く、診断の強力な手がかり |
| 筋緊張低下 | ≥75% | 新生児期から乳児期に顕著、運動発達遅滞の一因 |
| 特徴的顔貌 | ≥75% | 丸い顔、前頭部突出、短い人中、キューピッドの弓状口唇 |
| 言語発達遅滞 | 高頻度 | 表出言語の乏しさ、鼻声が特徴的 |
| 脳構造異常 | 30〜60% | 脳梁低形成・欠損、脳室拡大、髄鞘化遅延 |
| 先天性心疾患 | 一部の症例 | 心房中隔欠損、心室中隔欠損(HEY1欠失例で顕著) |
頭蓋顔面の特徴
本症候群の診断において、特徴的な顔貌(facial gestalt)の認識は極めて重要です。以下の特徴が75%以上の患者に共通して認められます。
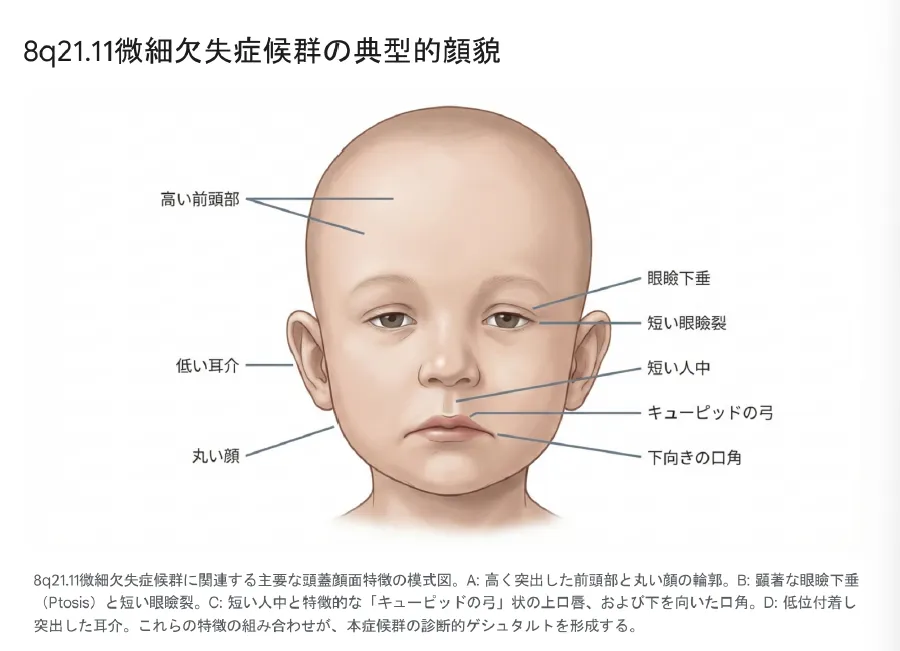
-
•
顔全体:丸い顔(Round face)、ふっくらとした頬、前頭部突出、短頭症
-
•
眼瞼部:両側性眼瞼下垂(Ptosis)、短い眼瞼裂、下向きの眼瞼裂
-
•
鼻・口唇部:広い鼻根部、短い人中、キューピッドの弓状上口唇、下がった口角
-
•
耳介・下顎:低位付着耳、突出耳、小顎症(Micrognathia)
眼科的異常とピーターズ奇形
眼の発生異常は8q21.11欠失症候群における主要な合併症であり、特にZFHX4遺伝子の欠失と強く関連しています。
💡 用語解説:「ピーターズ奇形」とは?
ピーターズ奇形(Peters Anomaly)は前眼部形成不全(Anterior Segment Dysgenesis)の一種で、角膜中央部の混濁、後部角膜実質・デスメ膜・内皮の欠損、および虹彩や水晶体と角膜の癒着を特徴とします。8q21.11欠失症候群は、ピーターズ奇形を伴う数少ない染色体異常の一つとして認識されています。
🔵 構造的異常
- •
ピーターズ奇形
- •
強膜化角膜(Sclerocornea)
- •
小眼球症、小角膜
- •
白内障、網膜色素変性
🔴 機能的異常
- •
斜視、眼振
- •
弱視(Amblyopia)
- •
高度の屈折異常
- •
大脳性視覚障害
神経学的および発達的特徴
中枢神経系の発達障害は必発であり、患者のQOLに最も大きな影響を与える要因の一つです。
-
①
知的障害:軽度〜中等度がほぼ全例で認められる
-
②
筋緊張低下:新生児期から乳児期に顕著、哺乳困難・摂食障害の原因に
-
③
運動発達遅滞:定頸、座位、歩行開始のマイルストーンが遅れる
-
④
言語発達遅延:表出言語の乏しさ、鼻声(口蓋機能不全関連)
-
⑤
行動特性:自閉スペクトラム症(ASD)様の行動、過度の親愛行動、睡眠障害
⚠️ 脳MRI所見:約30〜60%の患者に構造的異常が認められます。最も多いのは脳梁の低形成または欠損で、その他脳室拡大、脳回異常、髄鞘化の遅延、小脳虫部の低形成なども報告されています。
その他の全身合併症
❤️ 心血管系(HEY1欠失例)
- •
心房中隔欠損症(ASD)
- •
心室中隔欠損症(VSD)
- •
稀に左心低形成症候群(HLHS)
🦴 筋骨格系・四肢
- •
合指症、屈指症
- •
母指(趾)の幅広化
- •
先天性多発性関節拘縮
🔊 その他
- •
感音難聴
- •
停留精巣、小陰茎
- •
歯の萌出遅延

🩺 院長コラム【「三徴候」の重要性について】
8q21.11欠失症候群を疑う上で最も重要なのは、「知的障害・眼瞼下垂・筋緊張低下」の三徴候です。特に両側性の眼瞼下垂と丸い顔という顔貌の組み合わせは、経験のある臨床医であれば「何か染色体異常があるのでは」と直感的に感じる特徴です。
しかし、この疾患は100万人に1人未満という極めて稀な疾患であり、多くの医師が遭遇する機会がありません。「原因不明の発達遅滞」とされてきたお子さんの中に、実はこの症候群が隠れているケースがあると考えられます。
臨床遺伝専門医による診察と染色体マイクロアレイ検査を受けることで、診断に至る可能性があります。
3. 原因と遺伝的背景|責任遺伝子ZFHX4とHEY1
【結論】 本症候群の病態は、8q21.11領域に位置する複数の遺伝子のハプロ不全によって説明されます。特にZFHX4遺伝子は眼・神経発達の、HEY1遺伝子は心血管・神経学的特徴の主要なドライバーと考えられています。
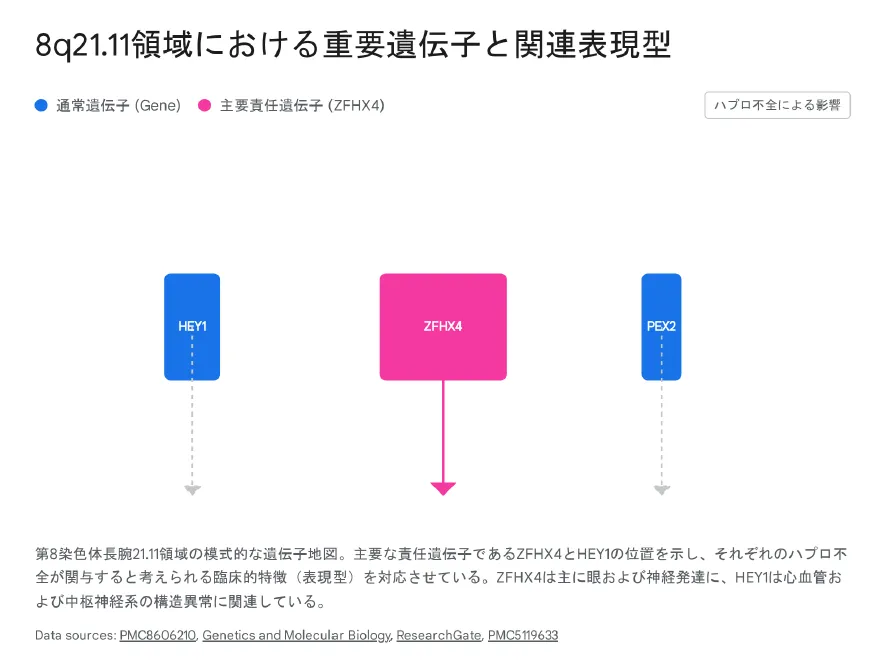
主要責任遺伝子の機能
| 遺伝子 | 主な機能 | 関連する臨床症状 |
|---|---|---|
| ZFHX4 | 転写因子(4つのホメオドメイン、22個のジンクフィンガー)、神経発生・分化の制御 | 眼瞼下垂、ピーターズ奇形、知的障害、小顎症 |
| HEY1 | Notchシグナル経路の転写因子、心臓・血管・中枢神経の発生 | 先天性心疾患、脳梁低形成、ASD様行動 |
| PEX2 | ペルオキシソーム形成因子 | 白内障、角膜混濁(ハプロ不全の影響) |
ZFHX4遺伝子:眼と神経発達の鍵
ZFHX4遺伝子は3,550個以上のアミノ酸からなる巨大な転写因子をコードしています。発生中の脳および筋肉組織で顕著に発現し、神経前駆細胞の運命決定や分化のタイミングを制御しています。
-
①
CHD4との相互作用:NuRD複合体と協調し、神経発生・軸索形成・神経細胞移動に関わる遺伝子を制御
-
②
ゼブラフィッシュモデル:zfhx4欠損でメッケル軟骨短縮・篩骨板小型化 → ヒトの小顎症・顔面異形成の機序を説明
-
③
運動機能への影響:zfhx4欠損ゼブラフィッシュは運動頻度低下 → ヒトの筋緊張低下と相関
HEY1遺伝子:心血管と神経の発達
HEY1遺伝子はNotchシグナル伝達経路の下流にある重要な転写因子です。心臓発生における上皮間葉転換(EMT)や心中隔形成を制御し、Hey1ノックアウトマウスは大動脈弓異常で周産期に死亡します。
心血管発生への影響
- •
心内膜の上皮間葉転換(EMT)制御
- •
心中隔の形成
- •
ASD、VSD、HLHSとの関連
神経学的機能への影響
- •
神経幹細胞の維持
- •
髄鞘関連遺伝子の発現制御
- •
ドーパミン輸送体(DAT1)制御 → ASD関連
遺伝子型-表現型相関
欠失領域に含まれる遺伝子によって、症状の組み合わせが異なることが明らかになっています。
| 臨床的特徴 | ZFHX4欠失あり | ZFHX4欠失なし(HEY1欠失あり) |
|---|---|---|
| 眼瞼下垂・眼球異常 | あり(+++) | なし(-) |
| ピーターズ奇形 | あり(++) | なし(-) |
| 知的障害 | あり(++) | あり(+) |
| 脳梁欠損・脳構造異常 | あり(+) | あり(++) |
| 心疾患(ASD/VSD) | あり(+) | あり(++) |
💡 ZFHX4は「眼症状と顔貌」、HEY1は「心血管と脳構造」の責任遺伝子
この比較から、ZFHX4は眼症状および顔貌の特徴の主要な責任遺伝子であり、HEY1は心血管および特定の脳構造異常により強く関連しているという仮説が支持されています。
欠失の発生機序
8q21.11領域の欠失は症例ごとに異なる箇所で起きた非反復的な構造変化です。低頻度反復配列(LCR)による反復的な欠失症候群ではなく、不均等な乗り換えよりも非アロイメント型のDNA二本鎖切断と再結合(NHEJなど)による可能性が高いと考えられています。
新生突然変異(多数例)
多くの症例は両親にその欠失がなく、孤発性(de novo)として発生します。配偶子形成時または受精後初期に新たに発生したものです。
家族性継承(稀)
稀に罹患した親からの常染色体優性(顕性)遺伝を示す家族例も確認されています。遺伝カウンセリングにおける重要な考慮事項です。
4. 8q21.11欠失症候群の診断方法
【結論】 本症候群の確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のG分染法では検出できない微細な欠失を高精度で検出できます。臨床症状のみで診断することはできません。
臨床診断のポイント
以下の臨床的特徴の組み合わせ(クラスター)が認められた場合、本症候群を鑑別疾患として考慮すべきです。
-
①
原因不明の知的障害および発達遅滞
-
②
先天性眼瞼下垂またはその他の眼前部異常(ピーターズ奇形など)
-
③
特徴的な顔貌(丸い顔、前頭部突出、キューピッドの弓状の上口唇)
-
④
筋緊張低下(新生児期〜乳児期)
遺伝学的検査の種類
| 検査方法 | 特徴 | 8q21.11欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 第一選択の診断ツール。ゲノム全域のCNVを高解像度で検出 | ◎ 確定診断可能 |
| G分染法(核型分析) | 解像度は5〜10Mb程度。大きな転座や数的異常を検出 | ✕ 検出困難(微小欠失のため) |
| 全エクソーム解析(WES) | ZFHX4遺伝子内の点変異を検出。CMA陰性でも症状がある場合 | △ ZFHX4点変異の検出に有用 |
| FISH法 | 特定領域のプローブを使用。両親の保因者診断に有用 | △ 確認検査として使用 |
💡 用語解説:染色体マイクロアレイ(CMA)とは?
CMAは、aCGH(アレイCGH)やSNPアレイを用いて、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。原因不明の発達遅滞・先天異常に対する第一選択検査として、日本でも保険適用で実施されています。
鑑別診断
類似した症状を呈する他の遺伝性疾患との鑑別が必要です。
歌舞伎症候群
知的障害、成長障害、特徴的顔貌を特徴。8q21.11とは眼瞼裂の形態が異なる(歌舞伎は長く、8q21.11は短い)
22q11.2欠失症候群
口蓋裂、心疾患、発達遅滞が共通。顔貌や免疫学的特徴(胸腺低形成)が異なる
8q22.1欠失症候群(隣接領域)
眼瞼下垂や顔貌の特徴が一部重複するが、CMAで明確に区別される
5. 治療と長期管理
【結論】 本症候群には根本的な遺伝子治療は存在しません。管理の主眼は、各臓器の合併症に対する対症療法・早期療育介入・定期的なサーベイランスに置かれます。多職種チームによる包括的アプローチが重要です。
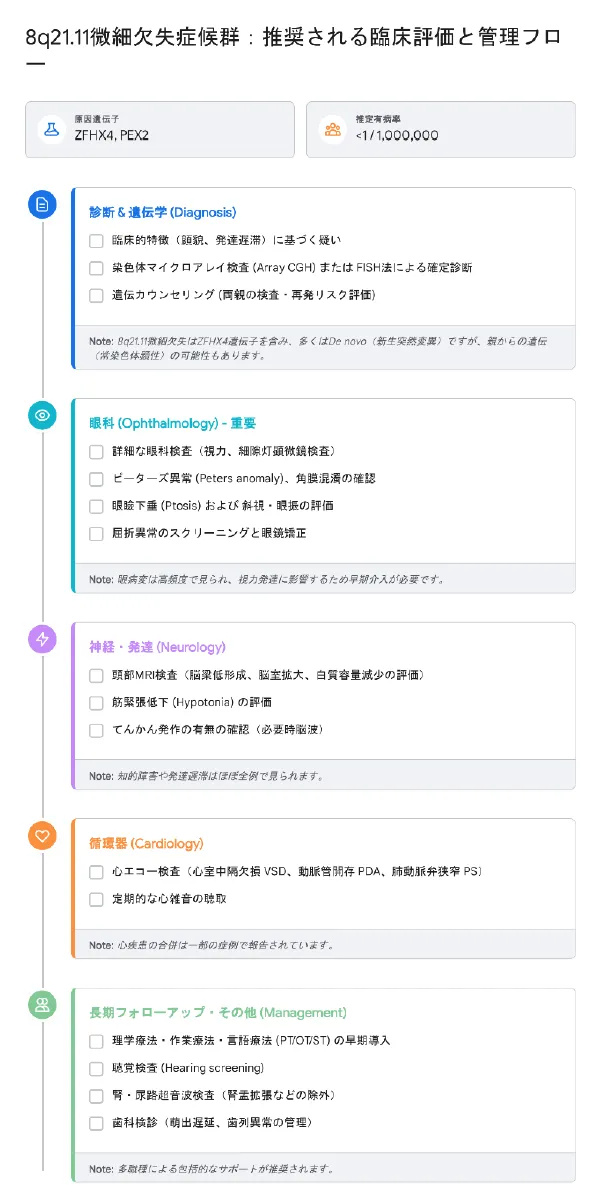
診断時に実施すべき評価項目
| 評価項目 | 内容 |
|---|---|
| 神経学的評価 | 詳細な神経診察、発達検査(認知、言語、運動) |
| 脳画像検査 | 頭部MRIによる脳梁欠損、脳室拡大、白質異常の有無の確認 |
| 眼科的評価(重要) | 視力、屈折、眼位、前眼部(角膜・水晶体)、眼底検査、眼瞼下垂の程度 |
| 循環器評価 | 心エコー検査および心電図による先天性心疾患のスクリーニング |
| 聴覚評価 | 聴性脳幹反応(ABR)や耳音響放射(OAE)を用いた聴力検査 |
| 腎・泌尿器評価 | 腹部超音波検査による腎奇形の有無、外性器診察(停留精巣) |
症状別の治療・対応
👁️ 眼科的管理
- •
屈折矯正(眼鏡装用)
- •
弱視治療(アイパッチ)
- •
重度眼瞼下垂には眼瞼挙上術
- •
ピーターズ奇形には角膜移植を検討
🧒 発達・教育的支援
- •
理学療法(PT):筋緊張低下に対応
- •
作業療法(OT):微細運動障害に対応
- •
言語聴覚療法(ST):言語遅滞・構音障害
- •
特別支援教育(IEP)の活用
❤️ 循環器・外科的管理
- •
先天性心疾患は循環器専門医と連携
- •
停留精巣は精巣固定術(1〜2歳頃)
- •
口蓋裂がある場合は形成外科的修復
-
•
臨床遺伝科:遺伝カウンセリング、家族への説明、長期フォロー
-
•
眼科:視力発達の管理、手術適応の判断
-
•
神経内科:てんかん発作への対応
-
•
小児循環器科:心疾患の評価・フォロー
-
•
リハビリテーション科:PT・OT・STの調整
予後とQOL
8q21.11欠失症候群の長期的な自然歴に関するデータは限られていますが、生命予後は一般的に良好と考えられています。ただし、左心低形成症候群(HLHS)のような重篤な心奇形を合併する場合は予後に影響する可能性があります。
✅ 早期療育の重要性:多くの患者さんは生涯にわたり何らかの支援を必要としますが、早期からの適切な療育介入により、コミュニケーション能力や日常生活動作(ADL)の獲得において良好な経過をたどるケースも報告されています。
6. 遺伝カウンセリングの重要性
【結論】 8q21.11欠失症候群の再発リスク評価のために、両親の遺伝学的検査が推奨されます。多くは新生突然変異ですが、稀に親からの遺伝もあるため、遺伝カウンセリングで正確な情報提供を受けることが重要です。
再発リスク
| 状況 | 次子への再発リスク | 備考 |
|---|---|---|
| 両親とも正常(新生突然変異) | <1% | 生殖細胞系列モザイクの可能性を完全否定できないため、ゼロではない |
| 親が均衡型転座保因者またはモザイク | 高い | 出生前診断や着床前診断(PGT)の選択肢を検討 |
| 罹患した患者本人から子へ | 50% | 常染色体優性(顕性)遺伝の形式に従う |
-
①
両親の検査:CMAまたはFISHで両親の保因者状態を確認
-
②
再発リスクの説明:新生突然変異(de novo)なら<1%、家族性なら高リスク
-
③
予後の不確実性:同じ欠失でも症状の重症度は個人差が大きい
-
④
出生前診断の選択肢:羊水検査・絨毛検査、着床前診断(PGT)の情報提供
🩺 院長コラム【遺伝カウンセリングで大切にしていること】
8q21.11欠失症候群のような稀少疾患の遺伝カウンセリングで最も重要なのは、「正確な情報」と「心理的サポート」のバランスです。
多くのご家族が「なぜうちの子だけ」という思いを抱えています。まずその気持ちに寄り添いながら、疾患の医学的情報、予想される経過、利用できる支援制度などを丁寧にお伝えしています。
臨床遺伝専門医として15年以上、医師になって以来、のべ10万人以上のご家族の意思決定と向き合ってきました。どのような決断をされても、その後もサポートを続けることをお約束しています。お一人で悩まず、ぜひご相談ください。
7. 出生前診断について
【結論】 8q21.11欠失は羊水検査・絨毛検査での染色体マイクロアレイ検査(CMA)により出生前に検出可能です。ただし、ミネルバクリニックのNIPT検査対象12種の微小欠失には含まれていませんので、確定検査としてのCMAが必要となります。
出生前検査での検出方法
| 検査 | 8q21.11欠失の検出 | 備考 |
|---|---|---|
| 羊水検査+CMA | ◎ 確定診断可能 | Gバンド法では検出できない微小欠失を確定診断可能。※学会指針では、原則として超音波での構造異常がある場合などが対象とされています。 |
| 絨毛検査+CMA | ◎ 確定診断可能 | 妊娠初期(11〜14週)に実施可能 |
| NIPT(12種微小欠失) | ✕ 対象外 | 8q21.11は対象12種(8q23q24 delとは別領域)に含まれない |
⚠️ NIPTの対象について:ミネルバクリニックで実施しているNIPTの12種微小欠失検査には8q23q24欠失が含まれますが、今回解説している8q21.11欠失は別の領域であり、NIPT検査の対象外です。本症候群のリスクが高い場合(上の子が罹患など)は、羊水検査・絨毛検査でのCMAによる確定検査をご検討ください。
出生前診断で見つかった場合の対応
出生前に8q21.11欠失が見つかった場合、超音波検査で半数以上で明らかな異常所見がないことが報告されています。そのため、予後予測は非常に難しくなります。
-
①
遺伝カウンセリング:欠失の意味、症状の範囲、予後の不確実性を説明
-
②
両親の検査:親が同じ欠失を持つか確認(家族歴の評価)
-
③
詳細超音波:心奇形、口蓋裂、眼球異常などの構造異常を精査
-
④
出生後フォロー体制:発達モニタリング、早期療育の準備
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。8q21.11欠失症候群を含む染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。全染色体検査や微小欠失検査も対応可能です。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会で費用面も安心
互助会制度(8,000円)により、NIPT陽性時の確定検査(羊水検査)費用が全額補助されます。上限なしで安心です。
一人で悩まず、専門医を頼ってください
8q21.11欠失症候群について詳しく知りたい方、
遺伝学的検査を検討している方は臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
関連記事







