目次
8p23.1微細欠失症候群とは?
症状・原因・診断・治療を臨床遺伝専門医が解説
Q. 8p23.1微細欠失症候群とはどのような病気ですか?
A. 第8染色体短腕(8p23.1)の約3.4〜5Mbが欠失することで、先天性心疾患・横隔膜ヘルニア・発達遅滞などを呈する染色体微細構造異常です。
この領域には心臓発生に重要なGATA4遺伝子が含まれており、そのハプロ不全が先天性心疾患(80〜90%)や横隔膜ヘルニア(20〜30%)の主因と考えられています。
-
➤
原因 → 8番染色体8p23.1領域(約3.4〜5Mb)の微細欠失 -
➤
主要症状 → 先天性心疾患(80〜90%)、発達遅滞(85%)、横隔膜ヘルニア(20〜30%) -
➤
重要な責任遺伝子 → GATA4(心臓発生)、SOX7、TNKS、MCPH1 -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
頻度 → 約1/50,000〜1/100,000出生(極めて稀な疾患)
1. 8p23.1微細欠失症候群とは|基本情報
【結論】 8p23.1微細欠失症候群(8p23.1 microdeletion syndrome)は、第8染色体短腕の8p23.1領域(約3.4〜5Mb)が欠失する染色体微細構造異常です。先天性心疾患を高頻度で合併することが最大の特徴であり、心臓発生のマスター転写因子であるGATA4遺伝子のハプロ不全がその主因です。
この症候群は隣接遺伝子症候群(contiguous gene syndrome)の一つであり、欠失領域に含まれる複数の遺伝子が半量になることで、心臓・横隔膜・神経発達など多臓器にわたる症状を呈します。1988年にFaganらによって初めて報告されて以来、世界中で症例が蓄積されています。
💡 用語解説:「隣接遺伝子症候群」とは?
染色体上で隣り合う複数の遺伝子が一緒に欠失(または重複)することで生じる症候群です。欠失した領域に含まれる各遺伝子のハプロ不全が組み合わさって、多彩な症状を呈します。8p23.1微細欠失症候群では、GATA4、SOX7、TNKS、MCPH1などの遺伝子が関与しています。
8p23.1微細欠失症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 8p23.1微細欠失症候群(8p23.1 microdeletion syndrome) |
| MedGen ID | C2931638 |
| 原因 | 8p23.1領域(REPD-REPP間)の約3.4〜5Mb欠失 |
| 頻度 | 約1/50,000〜1/100,000出生(Orphanetでは1〜9/100,000) |
| 遺伝形式 | 常染色体優性(顕性)遺伝(多くは新生突然変異) |
| 主要責任遺伝子 | GATA4、SOX7、TNKS、MCPH1、NEIL2 |
⚠️ 22q11.2欠失症候群との鑑別が重要
8p23.1微細欠失症候群は、22q11.2欠失症候群(DiGeorge症候群)と臨床像が類似することがあります。2023年のMontenegroらの研究では、22q11.2欠失症候群を疑われた心疾患児118例のうち8例(6.8%)が実は8p23.1欠失症候群であったと報告されています。心疾患+発達遅滞を呈する場合、両者の鑑別が重要です。
8p23.1領域のゲノム構造
8p23.1領域には、REPD(遠位反復配列)とREPP(近位反復配列)と呼ばれる低コピー反復配列(LCR)が存在します。これらの配列間で非対立遺伝子間相同組換え(NAHR)が生じることが、欠失発生の主なメカニズムです。
新生突然変異(約75〜95%)
- •
両親は正常で、配偶子形成時に新たに発生
- •
減数分裂時のNAHRが原因
- •
次子への再発リスクは低い(1%未満)
家族性継承(約5〜25%)
- •
片親が同じ欠失を保有
- •
不完全浸透のため親が無症状のことも
- •
次子への遺伝確率は50%
💡 用語解説:非対立遺伝子間相同組換え(NAHR)とは?
減数分裂時に、本来対応すべき位置ではなく、類似した配列(低コピー反復配列)同士で誤って組換えが起こる現象です。この結果、間に挟まれた領域が欠失したり重複したりします。8p23.1領域ではREPD-REPP間でNAHRが起こりやすく、これが本症候群の主要な発生メカニズムです。
2. 8p23.1微細欠失症候群の主な症状
【結論】 本症候群の最も重要な症状は先天性心疾患(80〜90%)であり、次いで発達遅滞(85%)、先天性横隔膜ヘルニア(20〜30%)が高頻度で認められます。ただし、症状の現れ方には個人差が非常に大きいことが特徴です。
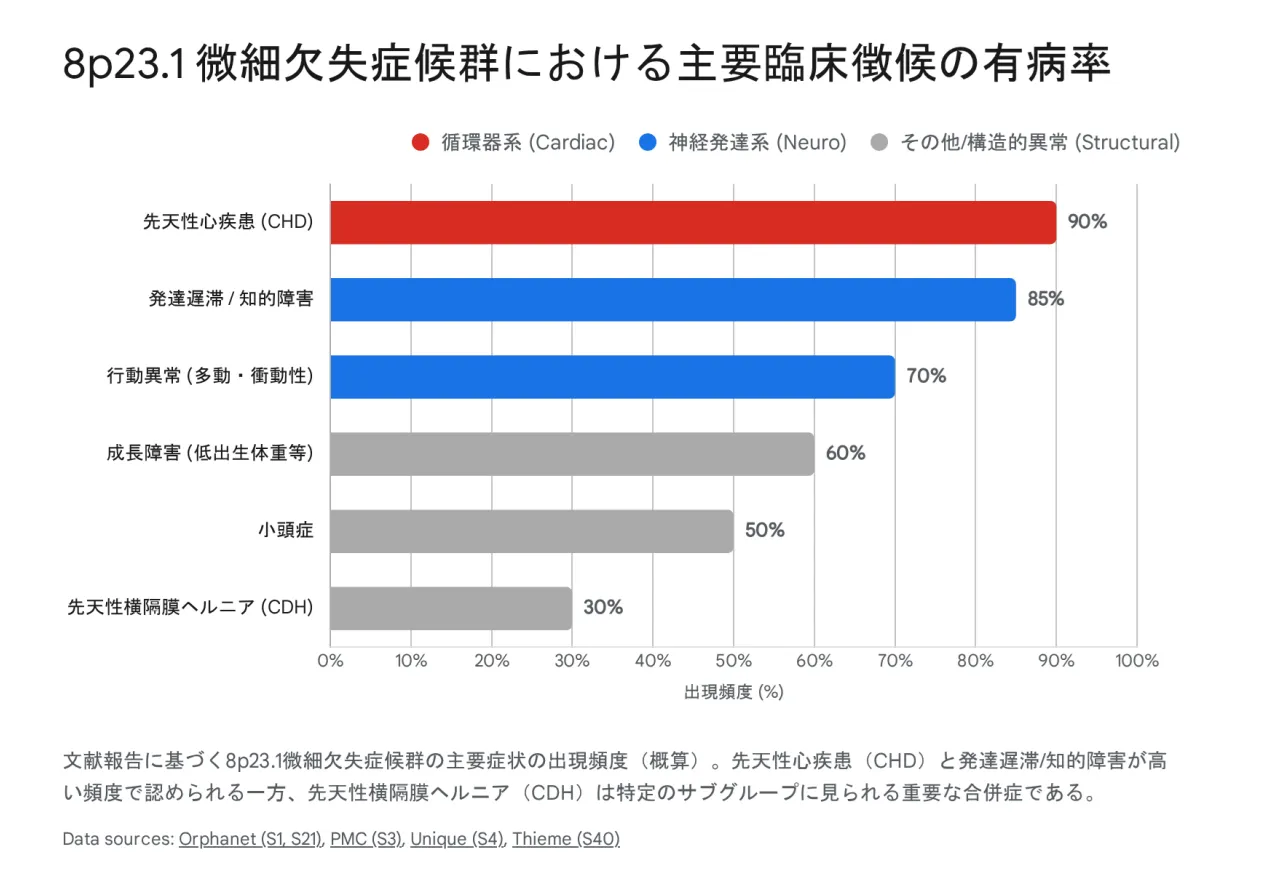
▲ 8p23.1微細欠失症候群における主要臨床徴候の有病率
症状の出現頻度
| 症状カテゴリー | 頻度 | 詳細 |
|---|---|---|
| 先天性心疾患 | 80〜90% | 心房中隔欠損(ASD)、心室中隔欠損(VSD)、房室中隔欠損(AVSD)、肺動脈狭窄、ファロー四徴症など |
| 発達遅滞・知的障害 | 85% | 軽度〜中等度が多い。まれに正常知能の報告も |
| 行動異常(多動・衝動性) | 70% | ADHD様症状、注意散漫、衝動性 |
| 成長障害 | 60% | 子宮内発育遅延(IUGR)、出生後の体重増加不良 |
| 小頭症 | 50% | 頭囲が小さい |
| 先天性横隔膜ヘルニア(CDH) | 20〜30% | 新生児期の呼吸不全の原因。緊急手術が必要 |
先天性心疾患(CHD):生命予後を左右する最重要合併症
先天性心疾患は本症候群の最も頻度が高く、かつ生命予後に直結する合併症です。心臓発生のマスター転写因子であるGATA4遺伝子のハプロ不全が主因と考えられています。
-
•
心房中隔欠損(ASD)・心室中隔欠損(VSD):最も高頻度。心内膜床由来の中隔形成不全
-
•
房室中隔欠損(AVSD):重度の心奇形。早期手術が必要
-
•
肺動脈弁狭窄:右室流出路の異常
-
•
ファロー四徴症・大血管転位:複雑心奇形。新生児期からの治療が必要
先天性横隔膜ヘルニア(CDH)
8p23.1欠失は症候性CDHの主要な遺伝的原因の一つです。横隔膜の一部が欠損し、腹腔内臓器が胸腔へ脱出することで、新生児期に重篤な呼吸不全を引き起こします。
⚠️ 重要:CDHは新生児救急疾患です。出生直後から呼吸管理が必要であり、状態が安定次第、外科的修復術を行います。8p23.1欠失が疑われる場合は、CDHの合併も念頭に置いた管理が重要です。
神経発達・行動特性
-
•
知的障害:軽度〜中等度が多い。正常知能の報告例もある
-
•
言語発達遅延:表出性言語(話す力)の遅れが顕著
-
•
ADHD様症状:多動性、衝動性、注意散漫(70%)
-
•
運動面:筋緊張低下、協調運動障害
身体的特徴(顔貌異常など)
本症候群の顔貌異常は比較的微細で非特異的であり、顔つきだけで診断することは困難です。
頭部・顔面
- •
小頭症
- •
小顎(下顎低形成)
- •
低位小耳症
- •
眼間開離
その他の身体所見
- •
口蓋裂・高口蓋(約10%)
- •
停留精巣・尿道下裂(男児)
- •
斜視・屈折異常
- •
てんかん(比較的稀)
💡 表現型の多様性と予測困難さ
興味深いことに、同じ欠失を持つ一卵性双生児でも、一方に重篤な横隔膜ヘルニアがあり、もう一方には見られなかったケースが報告されています。これは本症候群の表現型の多様性と予測困難さを示しており、遺伝カウンセリングを複雑にする要因となっています。

🩺 院長コラム【心疾患の早期発見と管理が予後を左右します】
8p23.1微細欠失症候群で最も重要なのは、先天性心疾患の早期発見と適切な管理です。80〜90%という高頻度で心疾患を合併するため、本症候群が疑われる場合は必ず心エコー検査を行います。
心疾患の種類は様々ですが、現代の小児心臓外科の発達により、適切な時期に適切な手術を行えば良好な予後が期待できます。新生児期の危機的状況を乗り越えれば、多くの患者さんは成人期まで生存可能です。
2023年のフランスの研究では、先天性心疾患が従来考えられていたより高頻度(17.3%ではなく、もっと高い)で見られることが確認されました。心疾患の精査は絶対に怠らないようにしています。
3. 原因と遺伝的背景|責任遺伝子の機能
【結論】 本症候群の原因は、8p23.1領域に位置するGATA4・SOX7・TNKS・MCPH1・NEIL2などの遺伝子のハプロ不全です。特にGATA4は心臓発生のマスター転写因子であり、そのハプロ不全が先天性心疾患と横隔膜ヘルニアの主因と考えられています。
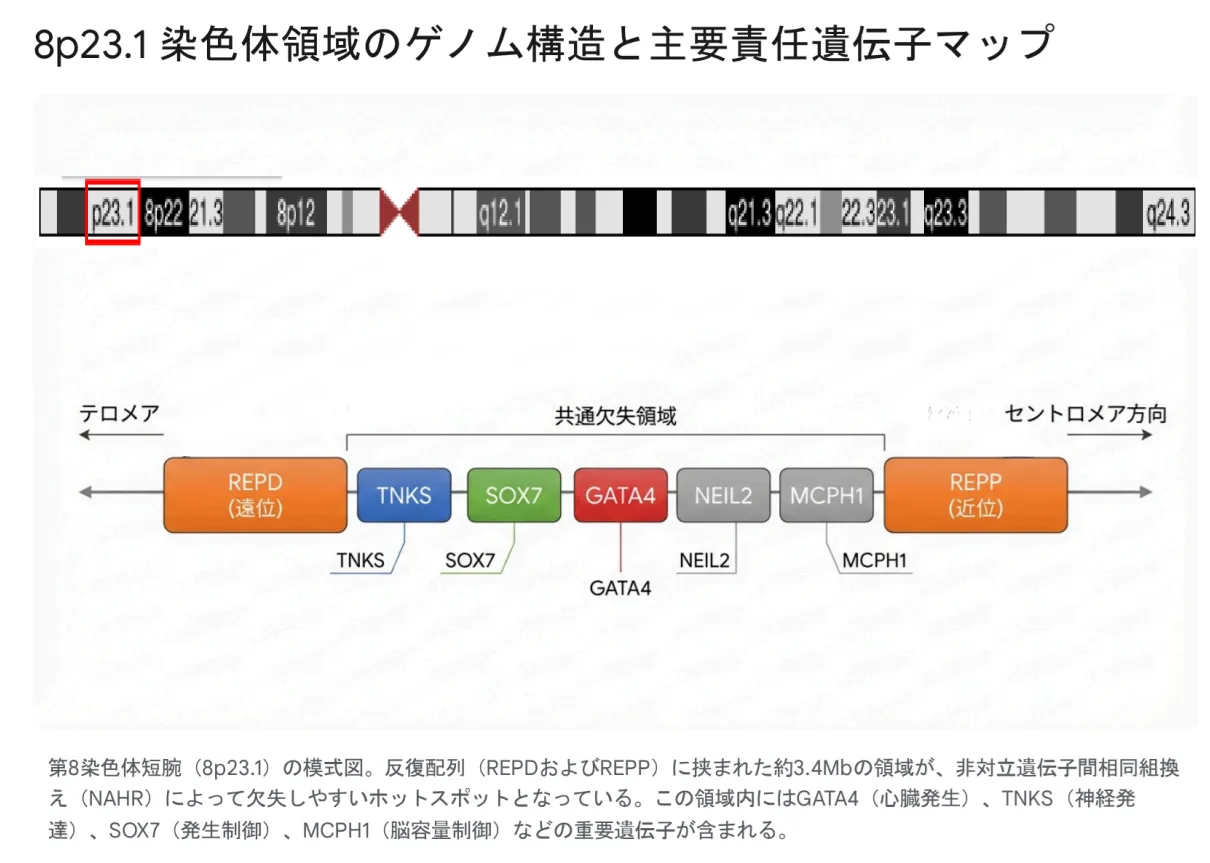
▲ 8p23.1染色体領域のゲノム構造と主要責任遺伝子マップ
💡 用語解説:「ハプロ不全」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。「ハプロ不全」とは、1コピーが欠失または機能しなくなることで、残り1コピーだけでは正常な機能を維持できない状態を指します。8p23.1欠失症候群では、欠失領域内の複数遺伝子がハプロ不全となることで多彩な症状が生じます。
主要責任遺伝子の機能
| 遺伝子 | 主な機能 | 関連する臨床症状 |
|---|---|---|
| GATA4 | 心臓発生のマスター転写因子。心筒形成、ループ形成、中隔形成を制御 | 先天性心疾患(ASD、VSD、AVSD)、横隔膜ヘルニア |
| SOX7 | GATA4と協調して心血管発生に関与。Wntシグナル、内皮間葉転換を制御 | 心疾患、横隔膜ヘルニア、発達遅滞 |
| TNKS | Wnt/β-カテニンシグナル調節、神経発達・シナプス可塑性に関与 | ADHD様行動、多動性、衝動性 |
| MCPH1 | 神経前駆細胞の分裂制御、脳容量決定、DNA損傷応答 | 小頭症、知的障害 |
| NEIL2 | 酸化損傷塩基の修復(DNAグリコシラーゼ) | 長期的な健康維持への影響(詳細は研究中) |
GATA4とSOX7:「二重ハプロ不全」仮説
興味深いことに、GATA4単独の点変異を持つ家系では家族性心疾患が報告されていますが、8p23.1欠失患者で見られる心疾患はより重篤で多様な傾向があります。
🎯 二重ハプロ不全仮説
GATA4とSOX7の両方が欠失することで、いずれか一方の欠失よりも重篤な心疾患や横隔膜ヘルニアが引き起こされるという仮説です。マウスモデルでも、Sox7欠失が心室中隔欠損を引き起こすことが示されており、この仮説を支持しています。
表現型の多様性を説明する要因
同じ欠失を持つ家族でも症状の程度が大きく異なる理由として、以下の要因が考えられています。
欠失サイズ・位置の違い
欠失の大きさや正確なブレークポイントは症例ごとに異なります。GATA4を含むかどうかで心疾患の有無が大きく変わります。
他の遺伝的修飾因子
残存する正常コピーの多型や、ゲノム全体の遺伝的背景が症状の重症度を修飾する可能性があります。
環境要因
胎内環境や出生後の環境因子が、遺伝的素因の発現に影響を与える可能性があります。
4. 8p23.1微細欠失症候群の診断方法
【結論】 本症候群の確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のG分染法(核型分析)では約3〜5Mbの微細欠失は検出困難なため、CMAが第一選択となります。
診断のきっかけ
-
①
先天性心疾患+発達遅滞:心疾患に加え、他の症状を合併する場合
-
②
先天性横隔膜ヘルニア:特に心疾患を合併している場合
-
③
原因不明の発達遅滞・知的障害:CMAが第一選択検査として実施される
-
④
22q11.2欠失症候群の除外:類似症状を呈するため鑑別が必要
-
⑤
出生前診断:胎児超音波で心奇形や発育不全が認められた場合
遺伝学的検査の種類
| 検査方法 | 特徴 | 8p23.1欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。数kb〜数Mbの微細CNVを高解像度で検出。欠失サイズ・ブレークポイントを正確に決定可能 | ◎ 検出可能 |
| G分染法(核型分析) | 解像度は5〜10Mb程度。大きな転座や数的異常を検出 | ✕ 検出困難(約3〜5Mbの微小欠失) |
| FISH法 | 8p23.1領域特異的プローブ(GATA4など)を使用。迅速な確認に有用 | ◎ 専用プローブで可能 |
| MLPA法 | 特定領域のコピー数を定量。比較的安価。22q11.2欠失との同時スクリーニングに有用 | ◎ 専用キットで可能 |
💡 用語解説:染色体マイクロアレイ(CMA)とは?
CMAは、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。SNPアレイやaCGH(アレイCGH)などの技術が用いられます。日本では原因不明の発達遅滞・先天異常に対する保険適用検査として実施されています。
鑑別診断
本症候群と臨床像が重複する疾患として、以下のものが挙げられます。
22q11.2欠失症候群
心疾患(特に円錐動脈幹異常)、口蓋裂、発達遅滞など多くの症状が共通。最も重要な鑑別疾患です。
8p23.1重複症候群
同一領域の重複。発達遅滞や軽度の異形を呈するが、表現型は欠失症候群とは異なります。
5. 治療と長期管理
【結論】 本症候群には根本的な治療法は存在せず、症状に応じた対症療法・早期療育・継続的支援が中心となります。特に先天性心疾患と横隔膜ヘルニアへの早期対応が生命予後を左右します。
ライフステージ別の管理
| ライフステージ | 主な対応 |
|---|---|
| 新生児期 | 心疾患・横隔膜ヘルニアの緊急対応、呼吸管理、栄養管理 |
| 乳児期・幼児期(0〜5歳) | 心臓手術、早期療育開始(PT・OT・ST)、発達評価、栄養サポート |
| 学童期(6〜12歳) | 知能検査・学習評価、特別支援教育の検討、ADHD症状への対応、行動療法 |
| 思春期・成人期 | 成人先天性心疾患(ACHD)フォロー、就労支援、生活自立支援、移行期医療 |
症状別の治療・対応
先天性心疾患
- •
心臓外科手術(中隔欠損閉鎖術など)
- •
内科的管理(強心薬、利尿薬)
- •
感染性心内膜炎予防
横隔膜ヘルニア
- •
新生児集中治療下での呼吸管理
- •
横隔膜外科的修復術
- •
術後の呼吸リハビリテーション
発達遅滞・言語障害
- •
早期療育が最も重要
- •
理学療法(PT)・作業療法(OT)
- •
言語聴覚療法(ST)
行動問題(ADHD様症状)
- •
行動療法・環境調整
- •
ソーシャルスキルトレーニング
- •
必要に応じてADHD薬物療法
-
•
臨床遺伝科:遺伝カウンセリング、家族への説明
-
•
小児循環器科:心疾患の評価・手術計画・長期フォロー
-
•
小児外科:横隔膜ヘルニア、口蓋裂などの手術
-
•
発達小児科:発達評価、療育調整
-
•
眼科・歯科:定期検診、必要な処置
予後
8p23.1微細欠失症候群の生命予後は、主に心疾患と横隔膜ヘルニアの重症度と管理状況に依存します。
📊 予後のポイント
- ✓
重篤な合併症に適切な治療を施せれば、成人期まで生存可能
- ✓
英国Uniqueのデータでは22歳の成人患者の報告あり
- ✓
出生時低体重でも、適切な栄養管理で身長・体重は正常範囲に達することが多い
- ✓
知的発達は個人差が大きく、正常知能の報告例もある
6. 遺伝カウンセリングの重要性
【結論】 8p23.1微細欠失症候群の表現型の多様性と不完全浸透は、遺伝カウンセリングを複雑なものにします。正確な情報提供と心理的サポートを通じて、ご家族の意思決定を支援することが重要です。
遺伝カウンセリングで伝えるべきポイント
-
①
症状の多様性:同じ欠失でも症状は無症状〜重度まで様々
-
②
不完全浸透:親が無症状で欠失を持っている可能性
-
③
予後の不確実性:現時点で正確な予後予測は困難
-
④
両親の検査:親が保因者か確認することで再発リスクを評価
-
⑤
長期フォローの必要性:多職種チームによる継続的支援が重要
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常(新生突然変異) | 1%未満(生殖細胞モザイクの可能性はあり) |
| 片親が保因者 | 50%(ただし症状発現は不確実) |
🩺 院長コラム【「予測できないこと」を正直に伝える】
8p23.1微細欠失症候群の遺伝カウンセリングで最も難しいのは、「予後が正確に予測できない」という不確実性をどう伝えるかです。
同じ欠失を持つ一卵性双生児でも症状が異なるケースがあるように、欠失の有無だけでは将来を予測することは困難です。心疾患が重篤な場合は早期の手術が必要ですし、逆に心疾患がなく発達も良好な例も報告されています。
私は臨床遺伝専門医として、「わかっていること」と「わかっていないこと」を正直にお伝えし、ご家族が自ら意思決定できるようサポートしています。どのような決断をされても、継続的にサポートすることをお約束します。
7. 出生前診断について
【結論】 8p23.1微細欠失症候群は出生前診断で検出可能です。羊水検査・絨毛検査でのCMAが確定診断のゴールドスタンダードですが、胎児超音波で心奇形や発育不全が認められた場合に検査が検討されます。
⚠️ ミネルバクリニックのNIPTでの検出について
ミネルバクリニックのNIPTで検出可能な微小欠失は12種類(1p36、2q33、4p16、5p15、8q23q24、9p、11q23q25、15q11.2-q13、17p11.2、18p、18q22q23、22q11.2)です。8p23.1微細欠失症候群はこの12種類には含まれておらず、確定診断には羊水検査・絨毛検査でのCMAが必要です。
出生前検査での検出方法
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| 胎児超音波検査 | △ 間接的 | 心奇形、横隔膜ヘルニア、発育不全などの所見から疑う |
| 羊水検査+CMA | ◎ 検出可能 | 確定診断のゴールドスタンダード |
| 絨毛検査+CMA | ◎ 検出可能 | 妊娠初期(11〜14週)に実施可能 |
出生前診断で見つかった場合の対応
-
①
遺伝カウンセリング:欠失の意味、表現型の多様性、予後の不確実性を説明
-
②
両親の検査:親が同じ欠失を持つか確認(保因者なら参考情報に)
-
③
詳細超音波・胎児心エコー:心奇形、横隔膜ヘルニアなどの精査
-
④
出生後フォロー体制:心臓外科、新生児科との連携準備
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。全染色体検査や微小欠失検査も対応可能です。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会で費用面も安心
互助会制度(8,000円)により、陽性時の確定検査(羊水検査)費用を全額補助。NIPT受検者全員に適用されます。
一人で悩まず、専門医を頼ってください
8p23.1微細欠失症候群について詳しく知りたい方、
出生前検査を検討している方は臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
🏥 一人で悩まないでください
8p23.1微細欠失症候群について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
参考文献
- [1] Orphanet. 8p23.1 microdeletion syndrome. [Orphanet]
- [2] NCBI MedGen. 8p23.1 microdeletion syndrome (Concept Id: C2931638). [MedGen]
- [3] Wat MJ, et al. Chromosome 8p23.1 Deletions as a Cause of Complex Congenital Heart Defects and Diaphragmatic Hernia. Am J Med Genet A. 2009;149A(9):2006-2012. [PMC]
- [4] Páez MT, et al. Two patients with atypical interstitial deletions of 8p23.1: Mapping of phenotypical traits. Am J Med Genet A. 2008;146A(9):1158-1165. [PMC]
- [5] Global Genes. 8p23.1 microdeletion syndrome. [Global Genes]
- [6] Montenegro WE, et al. Expanding the Phenotype of 8p23.1 Deletion Syndrome: Eight New Cases Resembling the Clinical Spectrum of 22q11.2 Microdeletion. J Pediatr. 2023;252:118-125. [PubMed]
- [7] Kaymak D, et al. Prenatal Diagnosis of 8p23 Deletion Syndrome by Single Nucleotide Polymorphism Microarray. J Fetal Med. 2021;8:97-101. [Thieme]
- [8] DECIPHER. 8p23.1 duplication syndrome. [DECIPHER]
- [9] Unique. 8p23 deletions. [Unique/RareChromo]
- [10] Zhan J, et al. SOX7 deficiency causes ventricular septal defects through its effects on endocardial-to-mesenchymal transition and the expression of Wnt4 and Bmp2. Hum Mol Genet. 2023;32(9):1460-1471. [PubMed]
関連記事







