目次
裂手裂足奇形1型(SHFM1)とは?
原因・症状・診断・治療を臨床遺伝専門医が解説
Q. 裂手裂足奇形1型(SHFM1)とはどのような病気ですか?
A. 第7染色体長腕(7q21.3-q22.1)の構造異常により、DLX5/DLX6遺伝子の機能不全が生じ、手足の中央部の形成不全を特徴とする先天性四肢奇形です。
「裂手裂足症」という名称の通り、手足の中央にV字型の裂け目が生じます。約35%で感音難聴を合併するのがSHFM1の大きな特徴です。
-
➤
原因 → 7q21.3-q22.1領域の欠失・転座・逆位などの構造異常 -
➤
主要症状 → 裂手裂足(中央列欠損)、合指症、感音難聴(約35%) -
➤
重要な特徴 → 不完全浸透率(30〜40%)と表現度の多様性 -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
頻度 → SHFM全体で約1/18,000〜90,000出生
1. 裂手裂足奇形1型(SHFM1)とは|基本情報
【結論】 裂手裂足奇形1型(SHFM1)は、第7染色体長腕(7q21.3-q22.1)の構造異常により、手足の中央部(中央列)が形成されない先天性四肢奇形です。約35%で感音難聴を合併するのが他のSHFM型との重要な鑑別点です。
裂手裂足奇形(Split-Hand/Foot Malformation: SHFM)は、手足の「中央列(Central rays)」の欠損または低形成を特徴とする先天性疾患群です。かつては特異な外見から「ロブスター・クロウ(Lobster-claw)変形」と呼ばれていましたが、現代では差別的な用語として使用が避けられています。
💡 用語解説:「中央列」とは?
手足の指は発生学的に「列(ray)」と呼ばれる単位で形成されます。中央列とは第2指(示指)、第3指(中指)、第4指(環指)に相当する領域を指します。SHFMではこの中央列が欠損し、手足の中央にV字型の裂け目が生じます。
SHFM1の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 裂手裂足奇形1型(OMIM #183600) |
| 別名 | SHFM1、Split Hand/Foot Malformation type 1、欠指症(Ectrodactyly) |
| 原因 | 7q21.3-q22.1領域の構造異常(欠失・転座・逆位) |
| 頻度 | SHFM全体で約1/18,000〜90,000出生 |
| 遺伝形式 | 常染色体優性(顕性)遺伝(不完全浸透) |
| 責任遺伝子 | DLX5、DLX6(機能的ハプロ不全) |
| 特徴的合併症 | 感音難聴(約35%)、モンディーニ奇形 |
SHFMの遺伝的分類
SHFMは遺伝的に高度な異質性(Genetic Heterogeneity)を有しており、少なくとも6つの型が知られています。
| 型 | 遺伝子座 | 原因遺伝子/変異 | 主な特徴 |
|---|---|---|---|
| SHFM1 | 7q21 | DLX5/DLX6(染色体再構成) | 感音難聴との関連が強い |
| SHFM2 | Xq26 | – | X連鎖性遺伝(1家系の報告) |
| SHFM3 | 10q24 | タンデム重複(325-570kb) | 頻度約20%、微細な重複変異 |
| SHFM4 | 3q27 | TP63 | EEC症候群など多数の症候群と関連 |
| SHFM5 | 2q31 | DLX1/DLX2(候補) | 欠指、低身長、口蓋裂、小眼球症など |
| SHFM6 | 12q13 | WNT10B | 常染色体潜性(劣性)遺伝 |
⚠️ 日本人における重要な鑑別疾患:BHLHA9関連疾患
日本人の遺伝的背景において特筆すべき点として、17p13.3領域(BHLHA9遺伝子)の重複が「創始者変異(Founder mutation)」として高頻度に認められます。臨床像がSHFM1に類似するため、日本の臨床現場ではBHLHA9関連疾患との鑑別が極めて重要です。
2. SHFM1の主な症状|四肢と聴覚への影響
【結論】 SHFM1の臨床像は四肢の形態異常と感音難聴(約35%)が中心です。ただし、同一家系内でも症状の重症度は大きく異なり、表現度の多様性が顕著です。
四肢の表現型
「裂手裂足」という名称が示す通り、手足の中央列の形成不全が中核的な症状です。
この分類は第1指間腔(母指と示指の間の空間)の状態に基づき、裂手の重症度を5段階で評価します。
-
Type I
正常な指間腔:第1指間腔は正常に保たれている。中央列の欠損のみで機能障害は軽度。
-
Type II
指間腔の狭小化:第1指間腔が狭くなっているが、母指と示指は分離している。
-
Type III
合指様の指間腔:第1指間腔が皮膚性合指のように癒着。骨は分離しているが皮膚でつながっている。
-
Type IV
指の融合:第1指間腔が消失し、母指と示指の骨が部分的に融合している。
-
Type V
指間腔の完全消失:第1指間腔が完全に欠損し、中手骨・指骨の著明な欠損を伴う。最重症型。
💡 臨床的意義:Type I・IIは把持機能が比較的保たれるため保存的治療も選択肢となりますが、Type III以上では外科的再建が必要となることが多いです。
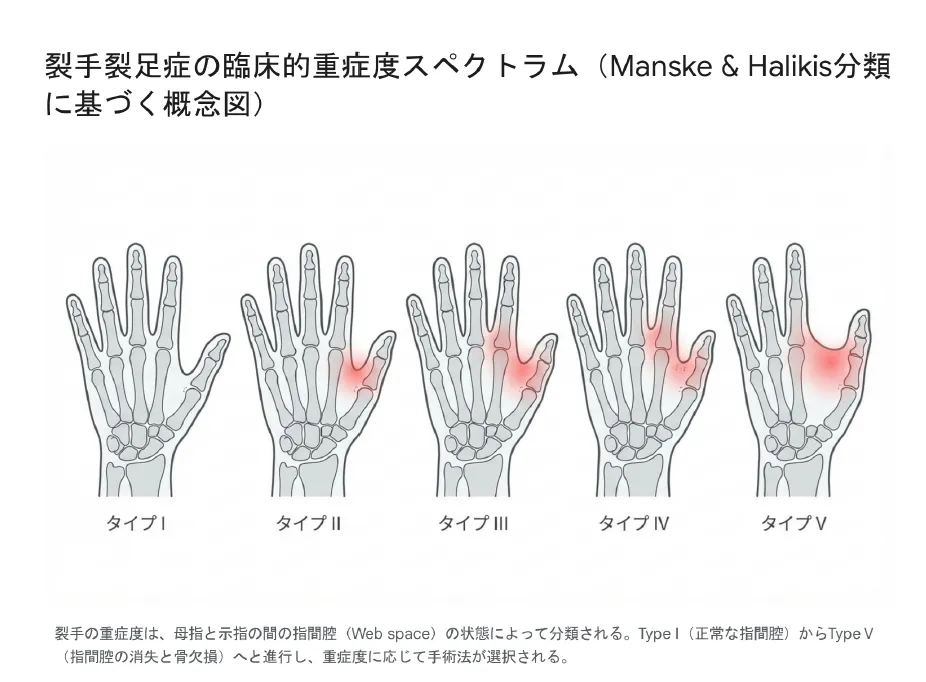
裂手の重症度分類:Type I(正常な指間腔)からType V(指間腔の消失と骨欠損)へと進行
典型的な形態異常
- •
中央列の欠損:第2〜4指の骨欠損
- •
V字型の裂け目(Cleft)の形成
- •
合指症:残存指の癒合
- •
三指節母指:母指の過剰分節
重症度のバリエーション
- •
軽症:軽微な合指のみ
- •
中等症:典型的な裂手裂足
- •
重症:単指症(1指のみ残存)
- •
無症状:不完全浸透例
聴覚障害:SHFM1を特徴づける合併症
SHFM1の診断において最も重要な臨床的指標が、感音難聴の合併(約35%)です。これは他のSHFM型との鑑別において強力な手がかりとなります。
-
•
タイプ:両側性の感音難聴(中等度〜重度)
-
•
内耳奇形:モンディーニ奇形(蝸牛1.5回転で発育停止、前庭水管拡大)
-
•
経過:先天性が多いが、幼児期〜学童期に進行するケースも
-
•
注意:新生児聴覚スクリーニングをパスしても継続的な経過観察が必要
💡 用語解説:モンディーニ奇形とは?
モンディーニ奇形は内耳の先天奇形で、蝸牛が正常な2.5〜2.75回転を形成できず、約1.5回転で発育が停止する状態です。DLX5/6遺伝子は耳胞(Otic Vesicle)の発生初期にも発現しており、内耳と四肢の両方の形態形成に必須であることが、SHFM1で難聴を合併する理由を説明しています。
その他の症状
SHFM1は「非症候群性」に分類されることが多いですが、一部の症例では以下の症状が報告されています。
| 症状 | 頻度 | 備考 |
|---|---|---|
| 感音難聴 | 約35% | SHFM1の特徴的な合併症 |
| 口唇口蓋裂 | 稀 | DLX遺伝子の鰓弓発生への関与を反映 |
| 知的障害・発達遅滞 | 稀 | 隣接遺伝子症候群の場合 |
⚠️ 重要:SHFM1患者の大多数で知能は正常です。知的障害を伴う場合は、染色体の微細欠失がSHFM1領域を超えて近傍の遺伝子に及んでいる可能性(隣接遺伝子症候群)を考慮する必要があります。
3. 原因と遺伝的背景|DLX5/DLX6遺伝子と機能的ハプロ不全
【結論】 SHFM1の原因は、7q21.3-q22.1領域に位置するDLX5・DLX6遺伝子の機能不全です。特筆すべきは、遺伝子のコーディング領域に変異がなくても、染色体構造異常によりエンハンサーとの連携が断たれると発症する「機能的ハプロ不全」のメカニズムです。
責任遺伝子:DLX5とDLX6
DLX5とDLX6は、ホメオボックス型転写因子をコードする遺伝子であり、四肢の発生において決定的な役割を果たします。
💡 用語解説:ホメオボックス(Homeobox)とは?
ホメオボックスは、約180塩基対からなる高度に保存されたDNA配列で、「ホメオドメイン」と呼ばれる60アミノ酸のDNA結合領域をコードしています。ホメオボックスを持つ遺伝子(ホメオボックス遺伝子)は転写因子として働き、他の多くの遺伝子の発現をオン・オフすることで、体の各部位が「どこに」「何を」形成するかを決定します。
ショウジョウバエで最初に発見され、ヒトを含むすべての動物で類似の遺伝子群が存在することから、進化的に極めて重要な「マスター制御遺伝子」として知られています。DLX5/DLX6は、四肢や内耳の形態形成を制御するホメオボックス遺伝子です。
💡 用語解説:外胚葉性頂堤(AER)とは?
外胚葉性頂堤(Apical Ectodermal Ridge: AER)は、発生中の肢芽(Limb Bud)の先端に形成されるシグナルセンターです。AERからのFGFシグナルが直下の中胚葉細胞の増殖を維持し、指の形成を可能にします。DLX5/DLX6はこのAERの維持と分化に不可欠であり、機能喪失によりAERが早期退縮すると中央列の形成不全(裂手裂足)が生じます。
| 遺伝子 | 主な機能 | 欠損時の影響 |
|---|---|---|
| DLX5 | AERの維持、内耳の形態形成、骨芽細胞分化 | 四肢形成不全、感音難聴 |
| DLX6 | DLX5と協調してAERを維持、Wntシグナルとの連携 | 四肢形成不全 |
| DSS1(SEM1) | プロテアソーム構成因子、DNA修復(BRCA2安定化) | 四肢への直接的影響は限定的 |
💡 マウスモデルからの知見
Dlx5またはDlx6のいずれか一方をノックアウトしたマウスでは明らかな四肢奇形は観察されませんが、両方を同時に欠損させたダブルノックアウトマウス(Dlx5/6 -/-)では、ヒトのSHFM1に酷似した典型的な裂手裂足が出現します。これは、DLX5とDLX6が機能的に重複(冗長性)していることを示しています。
機能的ハプロ不全のメカニズム
SHFM1の分子病態において最も注目すべき概念が、「機能的ハプロ不全」です。これは、原因遺伝子のコーディング領域自体に変異がなくても、その発現を制御するゲノム環境が破壊されることで発症するというメカニズムです。
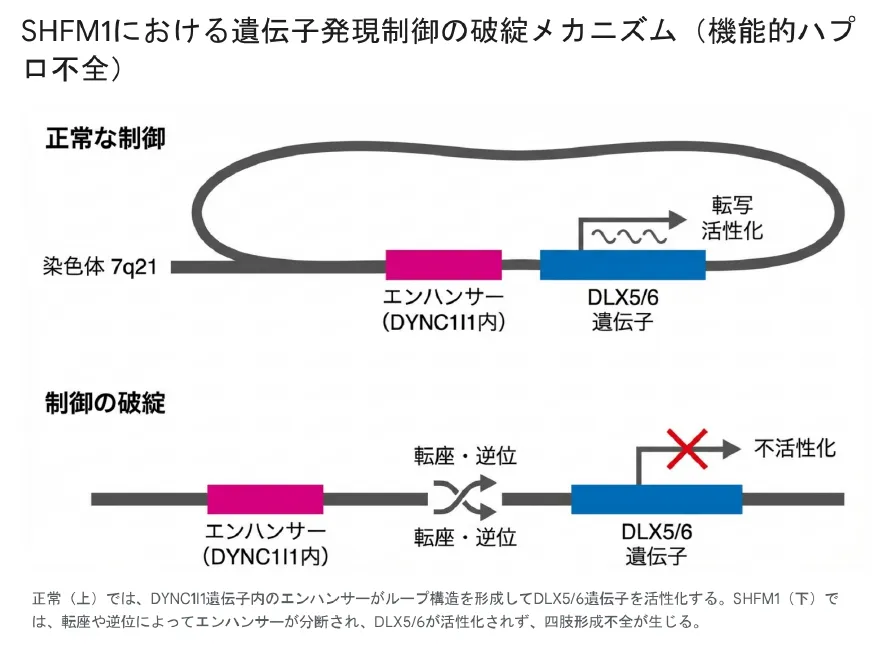
正常な制御(上)ではエンハンサーがループ構造を形成してDLX5/6を活性化。SHFM1(下)では転座や逆位によりエンハンサーが分断され、遺伝子が不活性化される。
💡 用語解説:エンハンサーとTAD
エンハンサーは、プロモーターから離れた場所に位置しながら遺伝子発現を活性化する制御配列です。DLX5/6の重要なエンハンサーは、DYNC1I1遺伝子のイントロン領域に存在することが判明しています。TAD(Topologically Associated Domains)は、これらの遠隔エレメントが相互作用する染色体上の区画です。
正常な状態
- ✓
DYNC1I1内のエンハンサーとDLX5/6が同一TAD内に存在
- ✓
クロマチンがループ状に折り畳まれ物理的に接近
- ✓
DLX5/6遺伝子が正常に転写される
SHFM1(転座・逆位)
- ✕
染色体切断点がエンハンサーとDLX5/6の間に位置
- ✕
エンハンサーが別の染色体領域へ移動
- ✕
遺伝子は正常でも転写が行われない
分子パスウェイのクロストーク:p63-DLX-Wnt軸
SHFM1の発症メカニズムは、四肢発生を司る広範な遺伝子ネットワークの破綻として理解する必要があります。
-
①
p63による上流制御:SHFM4の原因遺伝子TP63(p63タンパク質)は、DLX5/6エンハンサーに結合し発現を誘導する
-
②
Wntシグナルとの連携:DLX遺伝子群はWnt経路の構成要素として肢芽の成長を促進
-
③
BMPシグナルの制御:指間の細胞死(アポトーシス)や指骨の分化にも関与

🩺 院長コラム【「位置効果」が診断を難しくする理由】
SHFM1患者の遺伝子検査において、通常のサンガーシーケンスやエクソーム解析では原因が特定できない「原因不明例」が多いことが問題でした。これは、遺伝子のコーディング領域には異常がなく、染色体の立体構造の変化(転座・逆位)によってエンハンサーとの連携が断たれているためです。
この「位置効果(Position Effect)」による発症機序は、現代ゲノム医学の最先端トピックであり、構造変異解析(Structural Variant Analysis)の重要性を示しています。私のクリニックでは、こうした複雑な遺伝学的背景についても丁寧にご説明しています。
4. SHFM1の診断方法
【結論】 SHFM1の診断には、視診・X線検査による臨床評価に加え、染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダードです。特に難聴の合併はSHFM1を強く示唆する所見であり、聴覚評価も必須です。
臨床的・画像的アプローチ
-
①
視診:四肢形態の評価(中央裂、合指、欠指のパターン)
-
②
X線検査:中手骨・中足骨の欠損パターン、手根骨・足根骨の癒合確認
-
③
聴覚評価:ABR(聴性脳幹反応)、純音聴力検査
-
④
画像診断:難聴疑い時は側頭骨CT/MRIでモンディーニ奇形を確認
遺伝学的検査のフローチャート
| 検査 | 検出対象 | SHFM1の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 微細欠失・重複(CNV) | ◎ 第一選択検査 |
| G分染法(核型分析) | 転座・逆位・大きな構造異常 | ○ 構造異常の確認に有用 |
| NGSパネル/エクソーム | 点変異・小規模インデル | △ CMAで陰性の場合に検討 |
| 全ゲノムシーケンス(WGS) | 構造変異の切断点、非コード領域変異 | ◎ 機能的ハプロ不全の解明に有効 |
💡 用語解説:染色体マイクロアレイ検査(CMA)とは?
CMAは、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。SHFM1の主要な原因である7q21領域の微小欠失を高感度に検出できます。原因不明の発達遅滞・先天異常に対する第一選択検査として国際的に推奨されています。
⚠️ 日本人における診断戦略
日本人患者では、BHLHA9(17p13.3)重複がSHFMおよび長管骨低形成の主要な原因(創始者変異)です。臨床的には裂手裂足に加え脛骨などの長管骨低形成を伴うことが多いですが、典型的なSHFM1との鑑別は外見だけでは困難です。SHFM1の検索と並行してBHLHA9重複の有無を確認することが診断効率を高めます。
5. 治療と長期管理
【結論】 SHFM1の治療目標は機能とQOLの最大化です。四肢の形態異常には外科的再建、難聴には補聴器や人工内耳を含む包括的アプローチが必要です。多職種チームによる長期的サポートが不可欠です。
外科的治療:機能再建へのアプローチ
手の外科的治療は、把持機能(Pinch and Grasp)の獲得を最優先とし、整容的な改善を副次的な目的とします。手術時期は一般的に1歳前後から2歳の間に行われます。
裂手再建術式の比較:Snow-Littler法とMiura法

Snow-Littler法(左)は皮弁先端の血流不全リスクがある。Miura法(右)はより単純な皮弁デザインで血流を温存。
Snow-Littler法
- •
1967年に提唱された術式
- •
第1指間腔の拡大と裂の閉鎖を同時実施
- ⚠️
皮弁壊死・指間腔拘縮のリスク
Miura法(三浦法)
- •
1979年に三浦らが報告した改良法
- •
Z形成術応用の単純な皮弁デザイン
- ✓
血流温存、長期予後良好
聴覚・言語ケア
SHFM1患者の管理において、聴覚ケアは極めて重要です。
| 難聴の程度 | 推奨される対応 |
|---|---|
| 軽度〜中等度 | 補聴器装用、言語聴覚療法(ST) |
| 高度〜重度 | 人工内耳(Cochlear Implant)の検討 |
⚠️ 人工内耳手術の注意点:モンディーニ奇形などの内耳奇形を伴う場合でも人工内耳は有効ですが、術中に髄液噴出(Gusher)のリスクがあります。熟練した耳鼻咽喉科医による手術と慎重な術前計画が必要です。
多職種チームによる包括的ケア
-
•
臨床遺伝科:遺伝カウンセリング、家族への説明、再発リスク評価
-
•
整形外科・形成外科:裂手裂足の機能再建手術
-
•
耳鼻咽喉科:聴覚評価、補聴器・人工内耳
-
•
言語聴覚士(ST):言語発達支援
-
•
作業療法士(OT):手の機能訓練、日常生活動作支援
6. 遺伝カウンセリングの重要性
【結論】 SHFM1の不完全浸透率(30〜40%)と表現度の多様性は、遺伝カウンセリングを非常に複雑なものにします。家族への丁寧な説明と、不確実性を含めた情報提供が重要です。
遺伝形式と浸透率
| 遺伝形式 | 頻度 | 特徴 |
|---|---|---|
| 常染色体優性(顕性)遺伝 | 最多 | 罹患した親から50%の確率で伝達、不完全浸透(30〜40%) |
| 常染色体潜性(劣性)遺伝 | 稀 | SHFM1D(OMIM #220600):DLX5ホモ接合体変異など |
| 新生突然変異 | 一部 | 両親は正常、配偶子形成時に新たに発生 |
⚠️ 分離比の歪み(Segregation Distortion)
SHFM1では、罹患した父親から息子への伝達リスクがメンデル遺伝の予測値(50%)を有意に超える現象が報告されています。この生物学的メカニズムは未解明ですが、遺伝カウンセリングにおいて留意すべき点です。
カウンセリングで伝えるべきポイント
-
①
不完全浸透:変異を持っていても30〜40%は無症状
-
②
表現度の多様性:同一家系内でも症状の重症度は様々
-
③
「飛び石」遺伝:世代を飛ばした発症パターンがありうる
-
④
両親の検査:無症状の親が保因者かどうか確認することで再発リスクを評価
7. 出生前診断について|NIPTと羊水検査
【結論】 SHFM1は胎児超音波検査での四肢形態異常の発見をきっかけに診断されることがあります。羊水検査でのCMAが確定診断に有用ですが、不完全浸透と表現度の多様性により予後予測は困難です。
出生前検査での検出
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| 胎児超音波検査 | △ | 重度の四肢形態異常は妊娠中期以降に検出可能、軽症例は見逃されることも |
| NIPT | ✕ | SHFM1(7q21)はミネルバクリニックのNIPT対象外(12種類の微小欠失には含まれない) |
| 羊水検査+CMA | ◎ 確定診断 | Gバンド法では検出できない微小欠失を確定診断可能 |
⚠️ 羊水検査+CMAについて:学会指針では、原則として超音波での構造異常がある場合などが染色体マイクロアレイ検査(CMA)の対象とされています。胎児超音波で四肢形態異常が疑われた場合に実施を検討します。
⚠️ ミネルバクリニックのNIPTで検出可能な12種類の微小欠失
1p36 del、2q33 del、4p16 del、5p15 del、8q23q24 del、9p del、11q23q25 del、15q11.2-q13 del、17p11.2 del、18p del、18q22q23 del、22q11.2 del
※SHFM1(7q21)はこの12種類には含まれていません。胎児超音波で四肢形態異常が疑われた場合は、羊水検査でのCMAによる確定診断が必要です。
🩺 院長コラム【出生前診断で見つかった場合の対応】
胎児超音波で裂手裂足が疑われた場合、ご家族は大きな不安を抱えられることと思います。私のクリニックでは、まず正確な情報提供を心がけています。
SHFM1には「不完全浸透」と「表現度の多様性」があり、症状の程度を出生前に予測することは困難です。羊水検査で7q21領域の異常が確認されても、「どのような症状が出るか」は生まれてみないとわかりません。
この不確実性の中でご家族がどのような決断をされても、私は臨床遺伝専門医として最後までサポートします。不安なことがあれば、いつでもご相談ください。
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。SHFM1を含む染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。微小欠失12種類を含む包括的な出生前スクリーニングを提供しています。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会制度で費用面も安心
互助会制度(8,000円)により、NIPT陽性時の確定検査(羊水検査)費用が全額補助。上限なしで安心です。
一人で悩まず、専門医を頼ってください
裂手裂足奇形について詳しく知りたい方、
出生前検査を検討している方は臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
🏥 一人で悩まないでください
裂手裂足奇形について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
参考文献
- [1] Sowińska-Seidler A, et al. Split-hand/foot malformation – molecular cause and implications in genetic counseling. J Appl Genet. 2014;55(1):105-115. [PMC]
- [2] Ullah A, et al. Nonsyndromic Split-Hand/Foot Malformation: Recent Classification. Mol Syndromol. 2019;10(5):243-254. [PMC]
- [3] Delgado S, Velinov M. 7q21.3 Deletion involving enhancer sequences within the gene DYNC1I1 presents with intellectual disability and split hand-split foot malformation with decreased penetrance. Mol Cytogenet. 2015;8:37. [PMC]
- [4] Robledo RF, et al. The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev. 2002;16(9):1089-1101. [PubMed]
- [5] Lo Iacono N, et al. Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development. 2008;135(7):1377-1388. [Development]
- [6] Klopocki E, et al. A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J Med Genet. 2008;45(6):370-375. [PubMed]
- [7] Nakashima M, et al. Japanese founder duplications/triplications involving BHLHA9 are associated with split-hand/foot malformation with or without long bone deficiency and Gollop-Wolfgang complex. Orphanet J Rare Dis. 2014;9:125. [PubMed]
- [8] OMIM #183600 – Split-Hand/Foot Malformation 1; SHFM1. [OMIM]
- [9] GeneReviews – Split-Hand/Foot Malformation Overview. [GeneReviews]
- [10] Snow JW, Littler JW. Surgical treatment of cleft hand. Trans Int Soc Plast Surg 4th Congr. 1967;888-893.
- [11] Miura T. An appropriate treatment for postoperative Z-formed cleft hand. J Hand Surg Am. 1979;4(4):365-369. [PubMed]
関連記事







