目次

Q. 16p13.11微小欠失症候群とはどのような病気ですか?
A. 16番染色体短腕(16p13.11)の約0.8〜3.3Mb領域が欠失することで、神経発達障害や精神疾患のリスクが高まる染色体微細構造異常です。
NDE1・MYH11・NOMO1・NTAN1などの責任遺伝子のハプロ不全により、発達遅滞・てんかん・ASD・知的障害・統合失調症などの症状が生じることがあります。
-
➤原因 → 16番染色体短腕13.11バンドの約0.8〜3.3Mbの微小欠失
-
➤主要症状 → 発達遅滞(約90%)、てんかん(約60%)、小頭症(約50%)、ASD(約45%)
-
➤重要な特徴 → 不完全浸透率:欠失があっても症状は無症状〜重度まで様々
-
➤診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード
- ➤頻度 → 一般集団で約1/2,000〜3,000人(てんかん患者群では約0.5%)
1. 16p13.11微小欠失症候群とは|基本情報
【結論】 16p13.11微小欠失症候群は、16番染色体短腕13.11バンドの約0.8〜3.3Mb領域が欠失する染色体微細構造異常です。従来のG分染法では検出できないサイズのため「微小欠失」と呼ばれ、染色体マイクロアレイ検査(CMA)の普及により発見が増加しています。
「お子さんの発達が気になる」「検査で16p13.11欠失が見つかった」という方は、この病気について正確な情報を知ることが大切です。この症候群は「リスク因子(感受性因子)」として働き、欠失があるからといって必ず症状が出るわけではありません。
💡 用語解説:「不完全浸透率」と「表現度の差異」
遺伝学で「浸透率」とは、ある遺伝子変異を持つ人のうち実際に症状が現れる人の割合です。16p13.11欠失は不完全浸透(欠失があっても症状が出ない人がいる)かつ表現度の差異(症状が出る場合も軽度〜重度まで様々)を示すため、同一家系内でも症状の重篤度が大きく異なることがあります。
16p13.11微小欠失症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 16p13.11微小欠失症候群(MedGen: C4304596) |
| 原因 | 16p13.11領域の約0.8〜3.3Mb欠失 |
| 頻度 | 一般集団で約1/2,000〜3,000人(0.04〜0.05%) |
| 遺伝形式 | 常染色体優性(顕性)遺伝(不完全浸透) |
| 主要責任遺伝子 | NDE1、MYH11、NOMO1、NTAN1、ABCC1、ABCC6 |
| 発生機序 | 低コピー反復配列(LCR)間の非対立遺伝子間相同組換え(NAHR) |
欠失の発生メカニズム
16p13.11領域は、低コピー反復配列(LCRs:セグメンタル・デュプリケーション)が豊富に存在する構造的に不安定な領域です。減数分裂時にこれらの相同配列同士が誤って対合し、非対立遺伝子間相同組換え(NAHR)が起こることで欠失や重複が生じます。
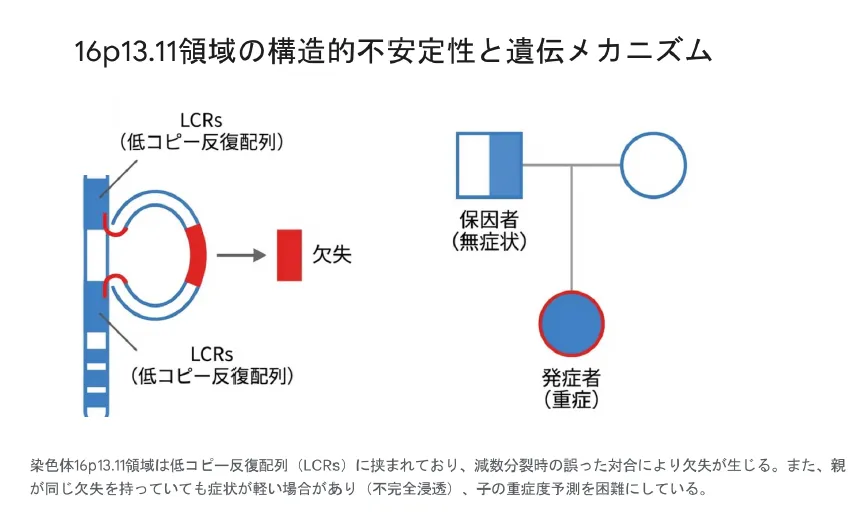
⚠️ 欠失と重複の違い
NAHRにより16p13.11領域には欠失(Deletion)と重複(Duplication)の両方が生じます。一般に欠失の方がハプロ不全による直接的な影響があるため、臨床的にはより重篤な表現型と関連する傾向があります。ただし重複も大動脈瘤などのリスク因子となります。
2. 16p13.11微小欠失症候群の主な症状
【結論】 本症候群の症状は発達遅滞(約90%)、てんかん(約60%)、小頭症(約50%)、ASD(約45%)、統合失調症(約20%)など多岐にわたります。ただし、不完全浸透率と表現度の差異により、同じ欠失でも症状は無症状〜重度まで様々です。
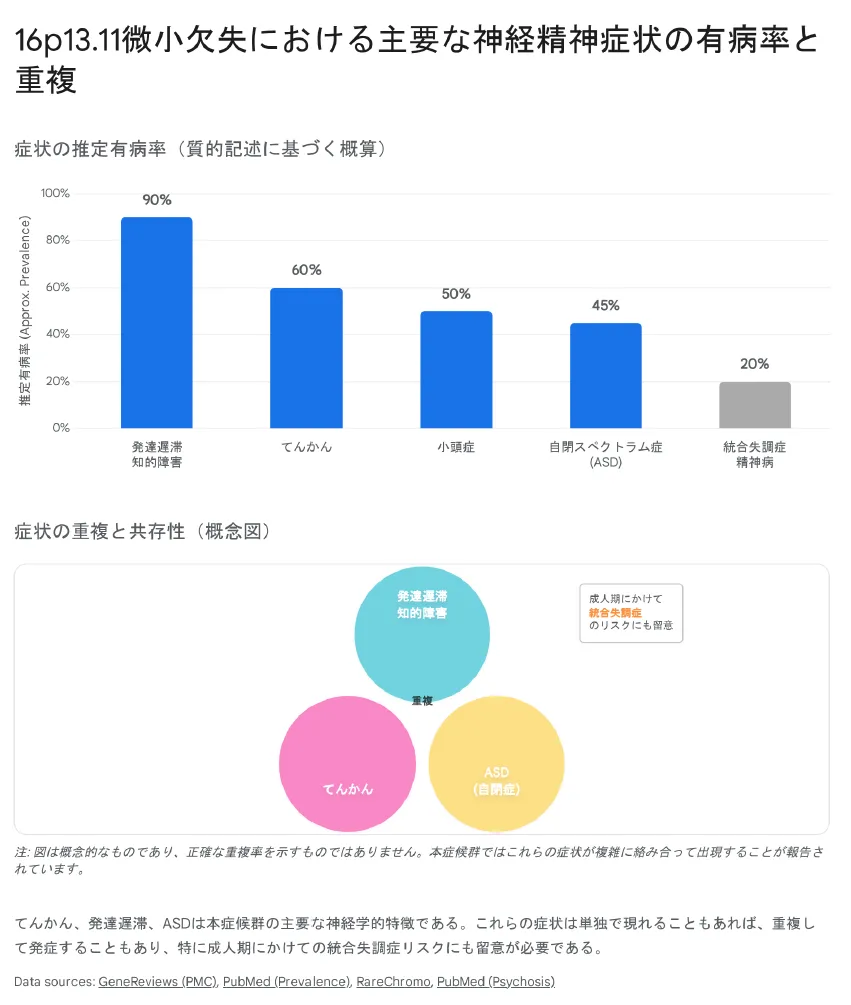
症状の出現頻度
以下の症状頻度は、症状があって検査を受けた患者群でのデータです。一般集団の保因者の中には無症状の方も多く存在することに注意してください。
| 症状カテゴリー | 頻度 | 詳細 |
|---|---|---|
| 発達遅滞・知的障害 | 約90% | 運動・言語発達遅延、IQ 80〜重度知的障害まで様々 |
| てんかん | 約60% | 点頭てんかん、欠神発作、全般強直間代発作など多様 |
| 小頭症 | 約50% | -2SD〜-3SD、NDE1欠失との強い関連 |
| 自閉スペクトラム症(ASD) | 約45% | 社会的コミュニケーション障害、限定的興味、常同行動 |
| ADHD | 約35% | 注意欠如、多動性、衝動性 |
| 統合失調症・精神病 | 約20% | 成人期発症、オッズ比2〜3倍 |
| 先天性心疾患 | 17〜30% | 心室中隔欠損、心房中隔欠損、ファロー四徴症など |
神経発達症状の詳細
- •
知的機能:IQ 80程度の境界域〜重度知的障害まで幅広いスペクトラム
- •
言語発達:表出性言語の遅れが顕著、3歳で単語レベルの例も
- •
運動発達:筋緊張低下、協調運動障害、運動失調
- •
行動面:爆発的な怒り、攻撃性、自傷行為(思春期以降に顕著化する場合あり)
てんかんの特徴
本症候群は遺伝性てんかんのリスク因子として最も頻繁に同定されるCNVの一つです。てんかん患者の大規模コホート研究で繰り返し有意な関連が報告されています。
発作型(多様)
- •
点頭てんかん(West症候群)
- •
欠神発作、全般強直間代発作
- •
複雑部分発作、ミオクロニー発作
- •
約1/3が薬剤抵抗性
推定メカニズム
- •
NDE1低下によるGABA作動性ニューロン遊走障害
- •
皮質回路の興奮・抑制バランス崩壊
- •
炎症関連遺伝子の発現変動
成人期の精神医学的リスク
本症候群の長期予後において成人期における精神疾患のリスクは見過ごされがちですが極めて重要です。
⚠️ 統合失調症リスク
16p13.11領域の欠失および重複は、統合失調症の強力な遺伝的リスク因子として確立されています。小児期・青年期早期の精神病症状や、てんかん発症後に幻覚・妄想を伴う「精神病性うつ病」を呈した成人例が報告されています。これは神経発達の初期段階におけるシナプス形成の微細な異常が、成熟後に精神病理として顕在化することを示唆しています。
身体的特徴
本症候群に特徴的な「顔」はありませんが、一部の患者さんでは以下のような所見がみられることがあります。
- •
頭部:微小脳症(-2SD〜-3SD)、短頭
- •
顔面:眼間開離、内眼角贅皮、鼻根部平坦化、薄い上口唇
- •
口蓋:高口蓋、口蓋裂
- •
四肢:多指症、合指症、彎曲指、関節弛緩性
- •
消化器:哺乳困難、胃食道逆流症(GERD)、慢性便秘

🩺 院長コラム【「感受性CNV」としての正しい理解】
16p13.11欠失について最も重要なことは、「欠失=病気」ではないということです。この欠失は「感受性CNV(Susceptibility CNV)」と呼ばれ、脳の発達に対する「バッファ(予備能)」を下げる因子として働きます。
症状が現れるかどうかは、他の遺伝的要因(Second Hit)や環境要因との組み合わせによって決まります。同一家系内でも、無症状の親から重度の発達障害を持つ子が生まれることがあり、これが「予後予測の困難さ」の原因です。
出生前診断や小児の発達検査で見つかった場合、この不確実性を遺伝カウンセリングで丁寧にお伝えしています。
3. 原因と遺伝的背景|責任遺伝子群の機能
【結論】 本症候群の病態は、16p13.11領域に含まれるNDE1・MYH11・NOMO1・NTAN1・ABCC1・ABCC6などの責任遺伝子のハプロ不全に加え、残存対立遺伝子の変異や他のゲノム変異との相互作用によって形成されると考えられています。
💡 用語解説:「ハプロ不全」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。「ハプロ不全(Haploinsufficiency)」とは、1コピーが欠失することで残り1コピーだけでは正常な機能を維持できない状態を指します。タンパク質量が約50%に減少し、細胞機能に影響が出ます。
主要な責任遺伝子の機能
| 遺伝子 | 主な機能 | 関連する臨床症状 |
|---|---|---|
| NDE1 | 中心体タンパク質、神経前駆細胞分裂・遊走制御 | 微小脳症、皮質形成異常(最重要) |
| MYH11 | 平滑筋ミオシン重鎖、血管・消化管機能 | 大動脈病変リスク、消化管機能障害 |
| NOMO1 | Nodalシグナル調節、左右軸決定 | ASD様症状(ゼブラフィッシュモデル) |
| NTAN1 | N-末端アスパラギンアミダーゼ、タンパク質分解 | 社会的行動変化、記憶・学習能力低下 |
| ABCC1/ABCC6 | ABCトランスポーター、物質輸送 | 結合組織脆弱性、血管石灰化リスク |
NDE1:脳発達の中枢
本症候群における神経学的表現型、特に微小脳症と皮質形成異常の形成において、NDE1遺伝子が最も重要と考えられています。
- ①
中心体に局在:細胞分裂時の紡錘体配向制御、細胞質ダイニンのリクルート
- ②
神経前駆細胞の分裂制御:対称性→非対称性分裂の移行を制御(大脳皮質ニューロン数を決定)
- ③
機能不全の影響:神経前駆細胞の早期枯渇、新生ニューロン遊走障害 → 微小脳症・皮質形成異常
「Two-Hit」仮説と表現型の多様性
同じ欠失を持つ家族でも症状の程度が大きく異なる理由として、「Two-Hit(2ヒット)仮説」が提唱されています。
🎯 Two-Hit仮説
第1ヒット:16p13.11微小欠失 → 脳の発達バッファ(予備能)を低下させる
第2ヒット:以下のいずれかが追加される
• 残存対立遺伝子の変異(特にNDE1の点変異)
• 別の染色体上のCNV(例:15q11.2欠失など)
• 環境要因(胎内感染、周産期トラブルなど)
→ 2つのヒットが重なることで閾値を超え、症状が顕在化
→ 第1ヒットのみ(無症状の親)では閾値を超えず発症しない
⚠️ NDE1の「Two-Hit」典型例:16p13.11欠失を持つ患者において、残存するもう一方の染色体上のNDE1遺伝子に微細な病的変異(点変異など)が存在する場合、NDE1機能が完全に喪失(Null状態)し、極めて重篤な微小脳症や脳形成不全(Lissencephaly type 4)を発症することが報告されています。
4. 16p13.11微小欠失症候群の診断方法
【結論】 本症候群の確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のG分染法では検出できない微細な欠失を高精度で検出できます。臨床症状のみで診断することはできません。
診断のきっかけ
- ①
原因不明の発達遅滞・知的障害:CMAが第一選択検査として実施される
- ②
難治性てんかんの精査:遺伝性てんかんのリスク因子として検索
- ③
自閉スペクトラム症の精査:遺伝学的原因検索として
- ④
先天性心疾患+発達遅滞:複合所見の原因検索
- ⑤
出生前診断:羊水検査でのCMAで偶発的に発見
遺伝学的検査の種類
| 検査方法 | 特徴 | 16p13.11欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。数kb〜数Mbの微細CNVを高解像度で検出 | ◎ 確定診断可能 |
| G分染法(核型分析) | 解像度は5〜10Mb程度。大きな転座や数的異常を検出 | ✕ 検出困難(0.8〜3.3Mbの微小欠失) |
| FISH法 | 特定領域のプローブを使用。迅速な確認に有用 | △ 専用プローブで可能 |
| MLPA法 | 特定領域のコピー数を定量。比較的安価 | △ 専用キットで可能 |
💡 用語解説:染色体マイクロアレイ(CMA)とは?
CMAは、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。日本では原因不明の発達遅滞・先天異常に対する保険適用検査として実施されています。アレイCGH法やSNPアレイなどの技術が用いられます。
5. 治療と長期管理|ライフステージ別アプローチ
【結論】 本症候群には根本的な治療法は存在せず、症状に応じた対症療法・早期療育・継続的支援が中心となります。ライフステージに応じた包括的かつ学際的なアプローチが不可欠です。
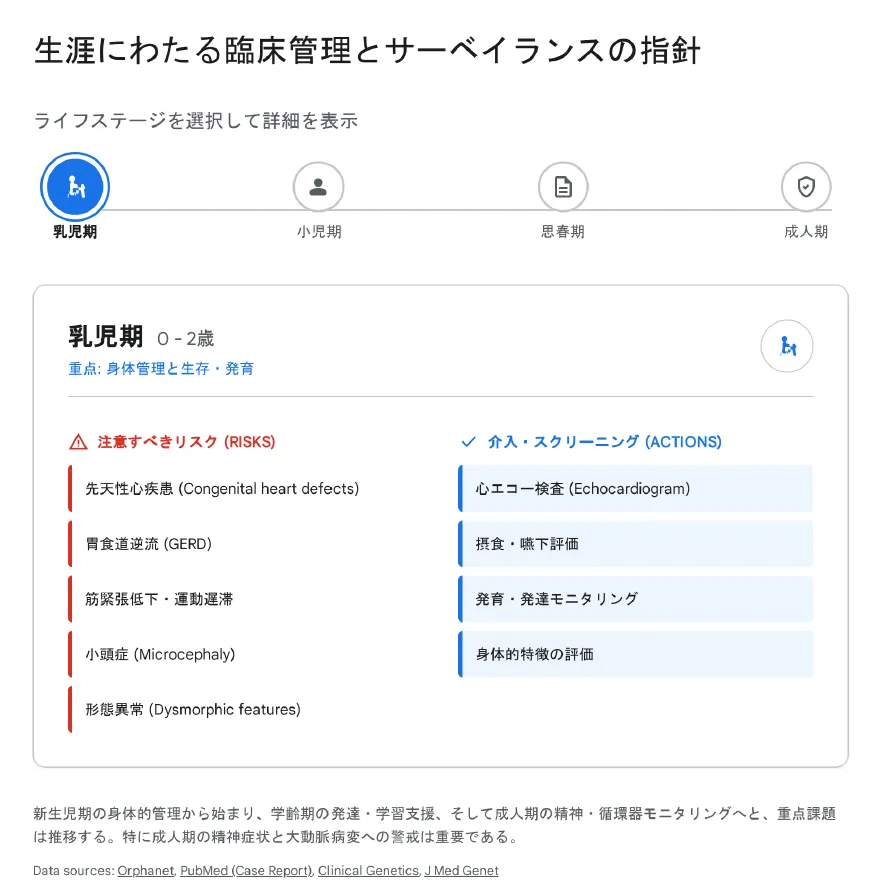
診断時の包括的評価(ベースライン)
診断が確定した時点で、症状の有無にかかわらず、全身のスクリーニングを行うことが推奨されています。
- •
遺伝学的検査:親の検査を行い、新生突然変異か遺伝性かを判定
- •
脳MRI:脳梁欠損、皮質形成異常、白質病変の確認
- •
脳波検査(EEG):てんかん波の有無をベースラインとして確認
- •
心エコー検査:先天性心疾患、大動脈基部径の測定
- •
腹部超音波:腎臓の形態確認(腎欠損、水腎症など)
- •
発達・心理検査:知能検査、適応行動検査、自閉症診断観察検査(ADOS-2)
ライフステージ別管理指針
| ライフステージ | 主な対応 |
|---|---|
| 乳児期(0〜2歳) | 哺乳困難・GERD対応、点頭てんかん監視、早期療育開始(PT) |
| 幼児・学童期(3〜12歳) | 言語療法(ST)・作業療法(OT)、特別支援教育の検討、てんかん・ADHD管理 |
| 思春期・青年期(13〜18歳) | 精神病前駆症状の監視、行動問題への対応、肥満予防 |
| 成人期(19歳〜) | 就労支援、精神疾患スクリーニング、大動脈径の定期モニタリング(MYH11欠失例) |
症状別の治療・対応
発達遅滞・言語障害
- •
早期療育が最も重要
- •
理学療法(PT)・作業療法(OT)
- •
言語聴覚療法(ST)
- •
特別支援教育の利用
てんかん
- •
発作型に応じた抗てんかん薬
- •
バルプロ酸、レベチラセタム等
- •
定期的な脳波検査
- •
難治例ではてんかん専門医連携
ADHD・ASD
- •
行動療法・環境調整
- •
ソーシャルスキルトレーニング
- •
ADHD薬物療法
- •
ペアレントトレーニング
精神症状(成人期)
- •
前駆症状の早期発見が重要
- •
抗精神病薬・抗うつ薬
- •
精神科との継続的連携
6. 遺伝カウンセリングの重要性
【結論】 16p13.11欠失の不完全浸透率と表現度の差異は、遺伝カウンセリングを非常に複雑なものにします。「欠失=必ず発症」ではないこと、予後予測が困難であることを丁寧に説明し、家族の意思決定を支援することが重要です。
遺伝カウンセリングで伝えるべきポイント
- ①
「リスク因子」としての性質:欠失は疾患の「確定原因」ではなく「感受性因子」
- ②
不完全浸透率:欠失があっても無症状で生活している人も存在
- ③
予後の不確実性:同じ欠失でも症状は無症状〜重度まで様々
- ④
両親の検査:親が保因者か確認することで再発リスクを評価
- ⑤
長期フォローの必要性:成人期の精神疾患・大動脈疾患リスクへの備え
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常(新生突然変異) | 1%未満(生殖細胞系列モザイクの可能性はあり) |
| 片親が保因者 | 50%(ただし症状発現は不確実) |
⚠️ 重要:親が同じ欠失を持ちながら健康である場合、それは子どもの予後にとってある程度の安心材料になりますが、完全に同じ経過をたどる保証はありません。「Two-Hit仮説」により、子どもには親にはない別の遺伝的要因(Second Hit)がある可能性があります。
🩺 院長コラム【遺伝カウンセリングで大切にしていること】
16p13.11欠失の遺伝カウンセリングで最も難しいのは、「予後が予測できない」という不確実性をどう伝えるかです。「欠失があるから病気」でも「欠失があっても大丈夫」でもなく、「リスクは上がるが、発症するかどうか、どの程度の症状が出るかは現時点では予測できない」という事実を正直にお伝えしています。
特に出生前診断で見つかった場合、ご家族は大きな決断を迫られます。私は中立的な立場で正確な情報を提供し、最終的な判断はご家族自身に委ねます。どのような決断をされても、その後もサポートを続けることをお約束しています。
臨床遺伝専門医として、のべ10万人以上のご家族の意思決定と向き合ってきました。不安を抱えている方は、ぜひ一度ご相談ください。
7. 出生前診断について|NIPTと羊水検査
【結論】 16p13.11微小欠失は出生前診断で検出可能です。羊水検査でのCMAが確定診断となりますが、胎児の予後予測は困難であり、出生前診断で見つかった場合の対応には慎重な遺伝カウンセリングが必要です。
⚠️ 重要:16p13.11はNIPT検出対象外です
ミネルバクリニックのNIPTで検出可能な微小欠失は12種類(1p36, 2q33, 4p16, 5p15, 8q23q24, 9p, 11q23q25, 15q11.2-q13, 17p11.2, 18p, 18q22q23, 22q11.2)です。16p13.11はこのリストに含まれていません。
16p13.11欠失の確定診断には羊水検査でのCMA(染色体マイクロアレイ検査)が必要です。
出生前検査での検出方法
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| NIPT(微小欠失検査) | ✕ 検出対象外 | ミネルバの12 microdeletionsに16p13.11は含まれない |
| 羊水検査+CMA | ◎ 確定診断 | Gバンド法では検出できない微小欠失を確定診断可能 |
| 絨毛検査+CMA | ◎ 確定診断 | 妊娠初期(11〜14週)に実施可能 |
⚠️ 学会指針について:羊水検査でのCMA(染色体マイクロアレイ検査)は、学会指針では原則として超音波での構造異常がある場合などが対象とされています。詳しくは遺伝カウンセリングでご相談ください。
出生前診断での超音波所見
本欠失が出生前に疑われるきっかけとなる超音波所見には以下のものが報告されています。ただし、これらは非特異的であり、超音波検査では半数以上で明らかな異常所見がないことも報告されています。
- •
NT肥厚(胎児後頸部浮腫):妊娠初期のソフトマーカー
- •
高輝度腸管:腸管の輝度が骨と同程度に見える所見
- •
脈絡叢嚢胞:脳室内の嚢胞
- •
その他:羊水過多、胎児発育不全(IUGR)、単一臍帯動脈など
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
COATE法による高精度NIPTを提供。全染色体検査や微小欠失検査も対応可能です。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会で費用面も安心
互助会制度(8,000円)により、陽性時の確定検査(羊水検査)費用が全額補助されます。上限なしで安心です。
一人で悩まず、専門医を頼ってください
16p13.11欠失について詳しく知りたい方、
検査を検討している方は臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
🏥 一人で悩まないでください
16p13.11微小欠失症候群について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- [1] Ramalingam A, et al. 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet. 2011;56(7):541-544. [PubMed]
- [2] Heinzen EL, et al. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86(5):707-718. [PMC]
- [3] Ingason A, et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry. 2011;16(1):17-25. [PubMed]
- [4] Hannes FD, et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2009;46(4):223-232. [PubMed]
- [5] Bakircioglu M, et al. The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am J Hum Genet. 2011;88(5):523-535. [PubMed]
- [6] Guemez-Gamboa A, et al. Primary cilia in the developing and mature brain. Neuron. 2014;82(3):511-521. [PubMed]
- [7] Ullmann R, et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 2007;28(7):674-682. [PubMed]
- [8] RareChromo.org – 16p13.11 microdeletions. [RareChromo]
- [9] Orphanet – 16p13.11 microdeletion syndrome. [Orphanet]
- [10] NCBI MedGen – 16p13.11 microdeletion syndrome (C4304596). [MedGen]
関連記事






