目次

Q. 16p12.1微細欠失症候群とはどのような病気ですか?
A. 16番染色体短腕(16p12.1)の約520kbが欠失することで、神経発達障害のリスクが高まる染色体微細構造異常です。
この欠失の最大の特徴は「不完全浸透」と「Two-Hitモデル」です。欠失があっても多くは軽症〜無症状で、別の遺伝的要因(セカンド・ヒット)が加わることで重症化します。
-
➤
原因 → 16番染色体16p12.1領域(約520kb)の微小欠失 -
➤
主要症状 → 発達遅滞(95%)、言語障害(90%)、知的障害(70%)、先天性心疾患(20〜33%) -
➤
重要な特徴 → 約93%が親から遺伝(de novoは約7%のみ)、保因者の親は多くが無症状〜軽症 -
➤
Two-Hitモデル → 重症例の約24〜30%が別の大きなCNVを併せ持つ -
➤
頻度 → 発達遅滞児で約0.2〜0.3%(一般集団の約7倍)
1. 16p12.1微細欠失症候群とは|基本情報
【結論】 16p12.1微細欠失症候群(16p12.1 recurrent microdeletion syndrome)は、16番染色体短腕p12.1領域の約520kbが欠失する染色体微細構造異常です。この症候群の最大の特徴は「不完全浸透」であり、欠失を持っていても軽症〜無症状の方が多いという点にあります。
2010年、Girirajanらによる画期的な大規模研究により、発達遅滞や知的障害を有する児においてこの欠失が有意に高頻度で認められることが報告され、独立した疾患単位として確立されました。本症候群は、「Two-Hitモデル」という神経発達障害の遺伝的基盤を理解するための重要な概念を提唱した点でも、臨床遺伝学において極めて重要な意味を持ちます。
💡 用語解説:「不完全浸透」とは?
遺伝学で「浸透率」とは、ある遺伝子変異を持つ人のうち実際に症状が現れる人の割合です。16p12.1欠失は浸透率が低いため、欠失を持つ親(保因者)から子に遺伝しても、親は無症状で子が発症するというパターンが頻繁に観察されます。これが「不完全浸透」です。
16p12.1微細欠失症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 16p12.1反復性微細欠失症候群(OMIM #136570) |
| ゲノム座標 | chr16:21,937,124-22,419,483(GRCh38/hg38) |
| 欠失サイズ | 約520kb(キロベース) |
| 頻度(発達遅滞児) | 約0.2〜0.3%(オッズ比7.2、一般集団より有意に高い) |
| 遺伝形式 | 常染色体優性(不完全浸透) |
| 遺伝パターン | 約93%が親から遺伝、de novoは約7%のみ |
| 含まれる遺伝子 | EEF2K、CDR2、UQCRC2、POLR3E、VWA3A、PDZD9、MOSMO(7遺伝子) |
⚠️ 16p11.2欠失症候群との違い
16p12.1欠失と名前が似ている16p11.2欠失症候群は別の疾患です。16p11.2欠失は自閉症や肥満との関連が強く、より遠位(セントロメアから遠い側)に位置します。両者は異なる遺伝子セットを含み、臨床像も異なりますので、混同しないよう注意が必要です。
欠失の発生メカニズム
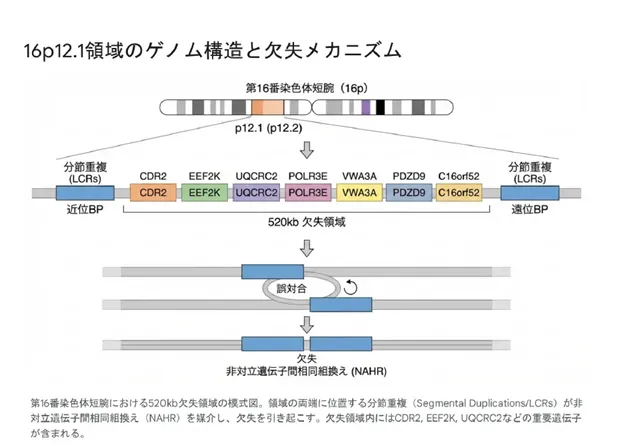
16p12.1欠失が発生する根本的な原因は、ヒトゲノムの進化過程で生じた「分節重複(Segmental Duplications)」の存在にあります。欠失領域の両端には約68kbの分節重複配列があり、これらが減数分裂時に非対立遺伝子間相同組換え(NAHR)を誘発します。
💡 用語解説:「分節重複(Segmental Duplications)」とは?
分節重複とは、ゲノム上の1kb以上の長さを持つDNA配列が、染色体の別の場所にも90%以上の高い相同性を保ったまま存在している構造です。ヒトゲノムの約5%は分節重複で構成されています。これらが近接して存在すると、減数分裂時に「誤った対合」が起こりやすくなり、NAHRによる欠失や重複の原因となります。
💡 用語解説:NAHR(非対立遺伝子間相同組換え)とは?
正常な減数分裂では、相同染色体上の同じ位置にある配列同士が対合します。しかし、16p12.1領域のように高い相同性を持つ重複配列が近接していると、「ずれた位置」で対合してしまうことがあります。この結果、一方の染色体では欠失、もう一方では重複が生じます。血縁関係のない世界中の患者でほぼ同一の切断点を持つ欠失が生じるのは、このNAHRが「反復的(Recurrent)」に発生するためです。
S2ハプロタイプ(82.4%)
- •
直列重複配列を含む
- •
NAHRを誘発しやすい
- •
欠失患者の99%がS2を持つ
S1ハプロタイプ(17.6%)
- •
重複配列が少ない構造
- •
欠失に対して保護的
- •
欠失発生リスクが低い
2. 16p12.1微細欠失症候群の主な症状
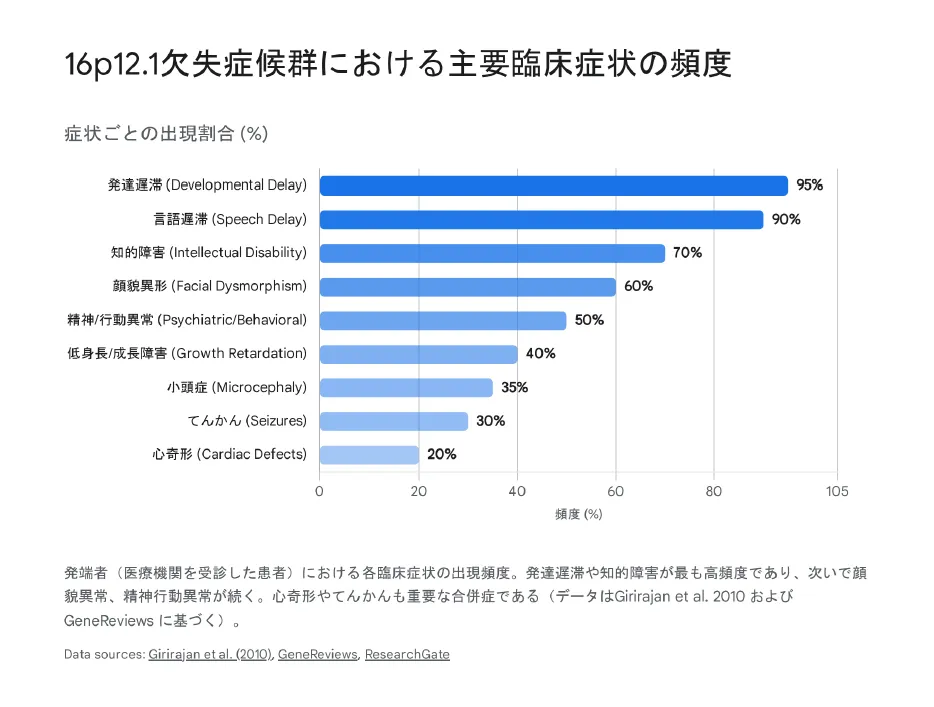
【結論】 本症候群の症状は発達遅滞(95%)、言語障害(90%)、知的障害(70%)、顔貌異常(60%)、先天性心疾患(20〜33%)、てんかん(30%)など多岐にわたります。ただし、不完全浸透率のため同じ欠失でも症状は無症状から重度まで様々です。
症状の出現頻度
以下の症状頻度は、症状があって検査を受けた患者群(発端者)でのデータです。保因者の親の多くは無症状〜軽症であることに注意してください。
| 症状カテゴリー | 頻度 | 詳細 |
|---|---|---|
| 発達遅滞 | 約95% | 全般的発達遅滞(運動・認知・社会性) |
| 言語発達遅延 | 約90% | 12ヶ月以上の全例で言語遅延(Girirajan et al.) |
| 知的障害 | 約70% | 軽度〜中等度が多い |
| 頭蓋顔面異形症 | 約60% | 軽微で非特異的、小顎症、耳介低位など |
| 精神・行動異常 | 約50% | ADHD、ASD、攻撃性、不安障害 |
| 成長障害 | 約40% | 低身長、胎内発育不全(SGA) |
| 小頭症 | 約35% | 頭囲-2SD以下 |
| 先天性心疾患 | 20〜33% | 左心低形成症候群(HLHS)、VSD、ASDなど |
| てんかん | 約30% | 点頭てんかん、欠神発作、全般強直間代発作など |
| 筋緊張低下 | 約48% | 乳児期からの低緊張、運動発達遅滞に寄与 |
神経発達症状の詳細
-
•
言語発達:表出性言語(発語)の遅れが顕著、2歳を過ぎても有意味語が出ないケースも
-
•
知的機能:軽度〜中等度の知的障害が多いが、境界知能域で学習障害を示す例も
-
•
運動発達:筋緊張低下により首座り、座位、独歩のマイルストーン達成に遅れ
-
•
行動面:ADHD、ASD的特徴、攻撃性、易刺激性(かんしゃく)、睡眠障害
精神医学的症状と成人期予後
本症候群では、成人期の精神疾患リスクも重要な特徴の一つです。
小児期〜思春期
- •
ADHD(注意欠如・多動性障害)
- •
自閉スペクトラム症(ASD)
- •
不安障害
- •
かんしゃく、攻撃性
成人期
- •
統合失調症(リスク上昇が報告)
- •
双極性障害
- •
重度のうつ病
- •
保因者の親にも軽度の精神症状
身体的特徴
本症候群に特徴的な「顔」はありませんが、軽微で非特異的な顔貌異常が報告されています。
-
•
顔面:小頭症、小顎症、深く窪んだ眼、低い鼻梁、眼間開離
-
•
耳部:耳介低位、耳介形成異常、耳前瘻孔
-
•
心臓:左心低形成症候群(HLHS)、心室中隔欠損、心房中隔欠損
-
•
その他:脊柱側弯症、手足の小奇形、停留精巣
⚠️ 重要:Girirajanらの初期研究では、21例中7例(33%)が先天性心疾患を持ち、そのうち4例が左心低形成症候群(HLHS)でした。HLHSは重篤な心奇形であり、本欠失が見つかった場合は心臓の精査が推奨されます。
3. Two-Hitモデル|なぜ同じ欠失でも症状が違うのか
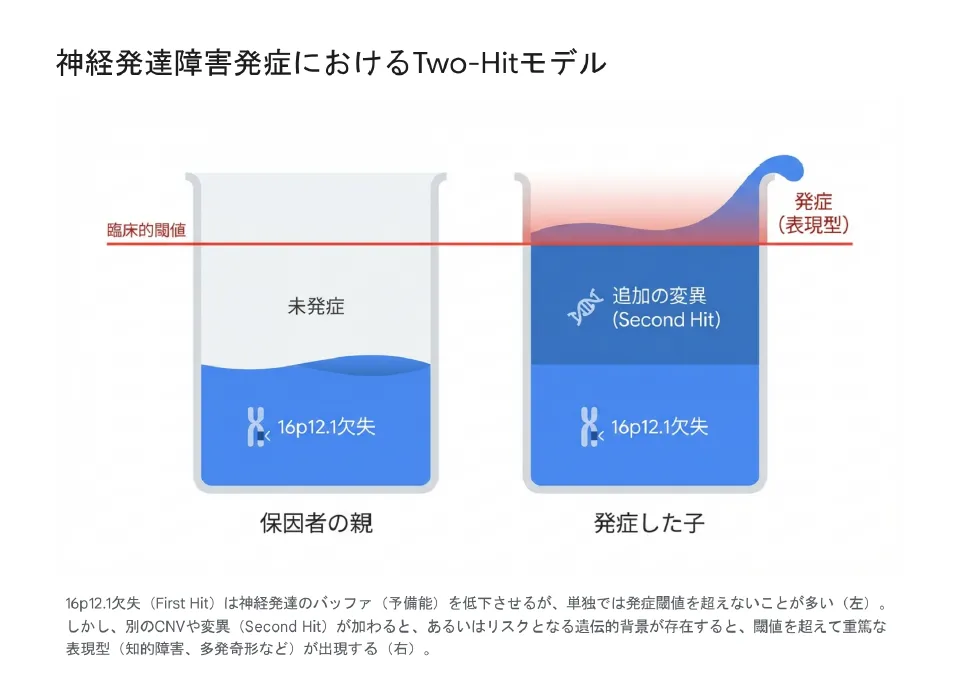
【結論】 16p12.1欠失症候群の最大の特徴は「表現型の可変性」と「不完全浸透」です。Girirajanらは、欠失を「ファースト・ヒット」とし、別の遺伝的要因(「セカンド・ヒット」)が加わることで重症化するという「Two-Hitモデル」を提唱しました。
ファースト・ヒット:16p12.1欠失の役割
16p12.1欠失は、神経発達に対する「バッファ(緩衝能)」を低下させる作用を持ちます。欠失があっても、ゲノムの他の領域が正常に機能していれば、この不安定性は補償され、臨床的な閾値を超える症状としては現れません。これが、欠失を持つ親(保因者)が無症状〜軽症である理由です。
セカンド・ヒット:閾値を超える要因
重度の発達遅滞や多発奇形などの顕著な表現型が現れる発端者においては、ファースト・ヒットに加えて、補償機構を破綻させる「セカンド・ヒット」が存在することが多いことがわかっています。
🎯 Two-Hitモデルの具体例
セカンド・ヒットの実体:
• 重症の発端者の約24〜30%で、16p12.1欠失以外に別の大きなCNV(>500kb)が検出
• 16p11.2、15q13.3、1q21.1などの神経発達症関連CNVが含まれる
相乗効果(Epistasis):
• ファースト・ヒットで脆弱になったシステムにセカンド・ヒットが加わると、影響が単なる足し算ではなく相乗的に増幅
• 発症閾値を超えて重篤な症状が出現
保因者の親(軽症〜無症状)
- •
16p12.1欠失のみ(ファースト・ヒット)
- •
「保護的」な遺伝的背景を持つ
- •
発症閾値を超えない
重症の発端者(子)
- •
16p12.1欠失+別のCNVや変異
- •
リスクを高める遺伝的背景を受け継ぐ
- •
発症閾値を超えて発症
💡 用語解説:ポリジェニック効果
明確な「大きなセカンド・ヒット」が見つからない症例でも、Two-Hitモデルの概念は適用可能です。この場合、セカンド・ヒットは単一の大きな変異ではなく、ゲノム全体に散在する多数の微細な変異(SNPs)の蓄積、すなわち「ポリジェニック・リスクスコア(PRS)」として機能していると考えられます。

🩺 院長コラム【Two-Hitモデルが示す重要な視点】
16p12.1欠失のTwo-Hitモデルは、「1つの遺伝子変異が1つの病気を作る」という単純な図式では捉えきれない、遺伝的背景の複雑さを示しています。
このモデルは本疾患だけでなく、自閉症、統合失調症、知的障害といった、ありふれた精神神経疾患の病態解明にも重要な示唆を与えています。16p12.1欠失は、個体の神経発達の堅牢性を揺るがす「感受性因子」であり、その最終的な表現型は、ゲノムの他の部分や環境との相互作用によって決定されるのです。
ご家族への説明では、「コップの水」の比喩を使うことがあります。「この欠失はコップの水を半分まで満たすものですが、それだけでは溢れません(発症しません)。他の遺伝的要因が積み重なって初めて水が溢れた(症状が出た)のです」と説明することで、単一の原因に帰結させない視点をお伝えしています。
4. 原因遺伝子|7つの責任遺伝子の機能
【結論】 16p12.1欠失領域には7つのタンパク質コード遺伝子(EEF2K、CDR2、UQCRC2、POLR3E、VWA3A、PDZD9、MOSMO)が含まれています。本症候群は、これら複数の遺伝子のハプロ不全が複合的に作用する「隣接遺伝子症候群」としての側面が強いです。
💡 用語解説:「ハプロ不全」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。「ハプロ不全」とは、1コピーが欠失または機能しなくなることで、残り1コピーだけでは正常な機能を維持できない状態を指します。タンパク質量が約50%に低下することで細胞機能に影響が出ます。
主要な責任遺伝子の機能
| 遺伝子 | 主な機能 | 関連する臨床症状 |
|---|---|---|
| EEF2K | タンパク質合成の「ブレーキ」役、シナプス可塑性に重要 | 知的障害、自閉的特性、心奇形リスク |
| CDR2 | 細胞周期の制御、小脳プルキンエ細胞で高発現 | 小脳機能障害(運動協調性低下、筋緊張低下) |
| UQCRC2 | ミトコンドリア電子伝達系複合体IIIの構成要素 | 神経発達遅延、易疲労性、筋緊張低下 |
| POLR3E | RNAポリメラーゼIII複合体のサブユニット | 頭蓋顔面異形症(小顎症、耳介変形) |
| MOSMO | Hedgehogシグナル伝達に関与 | 発生異常、顔面形成異常 |
EEF2K:神経発達における重要な役割
7遺伝子の中で、EEF2Kは神経精神症状の形成に特に重要と考えられています。
-
①
タンパク質合成制御:eEF2をリン酸化し、リボソームによるペプチド鎖伸長を一時停止
-
②
シナプス機能:NMDA受容体からのシグナルに応答、長期増強(LTP)/長期抑圧(LTD)に関与
-
③
ハプロ不全の影響:シナプスでのタンパク質合成脱抑制 → 神経回路形成異常
POLR3E:頭蓋顔面発生への関与
最近のXenopus(アフリカツメガエル)を用いた研究で、polr3eの発現低下が頭蓋顔面軟骨の形成不全を引き起こすことが示されました。これは、患者に見られる小顎症や耳介変形などの顔面異形症が、神経堤細胞の遊走・分化異常に起因することを示唆しています。
5. 16p12.1微細欠失症候群の診断方法
【結論】 本症候群は特異的な臨床症状に乏しいため、臨床診断(症状のみでの診断)は不可能です。確定診断には染色体マイクロアレイ検査(CMA)が必須となります。
診断のきっかけ
-
①
原因不明の発達遅滞・知的障害:CMAが第一選択検査として実施される
-
②
自閉スペクトラム症・ADHDの精査:遺伝学的原因検索として
-
③
てんかんの精査:特に難治性の場合
-
④
先天性心疾患+発達遅滞:複合所見の原因検索
-
⑤
出生前診断:羊水検査でCMAを行い偶発的に発見されることも
遺伝学的検査の種類
| 検査方法 | 特徴 | 16p12.1欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。数kb〜数Mbの微細CNVを高解像度で検出 | ◎ 確実に検出可能 |
| G分染法(核型分析) | 解像度は5〜10Mb程度。大きな転座や数的異常を検出 | ✕ 検出困難(約520kbの微小欠失) |
| FISH法 | 特定領域のプローブを使用。迅速な確認に有用 | △ 専用プローブで可能 |
| MLPA法 | 特定領域のコピー数を定量。比較的安価 | △ 専用キットで可能 |
💡 用語解説:染色体マイクロアレイ(CMA)とは?
CMAは、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。日本では原因不明の発達遅滞・先天異常に対する保険適用検査として実施されています。
親の検査(Parental Testing)の重要性
発端者で欠失が確認された場合、両親の検査を行うことが極めて重要です。
遺伝形式の確認
統計的には約93%の症例で親から遺伝しており、de novoは約7%のみです。親が保因者であれば再発リスクは50%と確定します。
セカンド・ヒットの検索
理想的には親子のトリオ解析を行い、発端者にあって親にはない他の遺伝子変異を探すことで、なぜ子が重症化したのかを説明できる場合があります。
6. 治療と長期管理
【結論】 本症候群には根本的な治療法は存在せず、症状に応じた対症療法・早期療育・継続的支援が中心となります。症状は多岐にわたるため、多職種チームによる包括的アプローチが重要です。
ライフステージ別の管理
| ライフステージ | 主な対応 |
|---|---|
| 乳児期・幼児期(0〜5歳) | 心エコー検査、発達スクリーニング、早期療育開始(PT・OT・ST)、聴力検査、眼科検査 |
| 学童期(6〜12歳) | 知能検査(WISC等)、特別支援教育の検討、ADHD・ASDへの対応、脊柱側弯症のチェック |
| 思春期・成人期(13歳〜) | 精神疾患の早期発見(統合失調症・うつ病)、就労支援、移行期医療、肥満管理 |

症状別の治療・対応
発達遅滞・言語障害
- •
早期療育が最も重要
- •
理学療法(PT)・作業療法(OT)
- •
言語聴覚療法(ST)
- •
代替コミュニケーション手段の導入
先天性心疾患
- •
診断時に心エコー検査を全例に
- •
小児循環器科での定期フォロー
- •
HLHSなど重症例は外科的治療
てんかん
- •
発作型に応じた抗てんかん薬
- •
定期的な脳波検査
- •
難治例ではてんかん専門医と連携
精神症状(成人期)
- •
前駆症状の早期発見が重要
- •
精神科との継続的連携
- •
睡眠・引きこもりに注意
7. 遺伝カウンセリングと出生前診断
【結論】 16p12.1欠失の不完全浸透率と表現型の多様性は、遺伝カウンセリングを非常に複雑なものにします。「欠失=必ず発症」ではないこと、予後予測が困難であることを丁寧に説明し、家族の意思決定を支援することが重要です。
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常(de novo) | 1%未満(生殖細胞モザイクの可能性はあり) |
| 片親が保因者 | 50%(ただし症状発現は不確実) |
出生前診断について
胎児超音波検査で心奇形や発育遅延が見つかった場合、羊水検査や絨毛検査でCMAを行い、この欠失が発見されることがあります。
⚠️ 出生前診断のジレンマ:出生前に診断された場合、表現型の予測が極めて困難であるため、両親への説明は慎重を要します。「欠失があっても健康に育つ可能性」と「障害を持つ可能性」の両方を、確率的な幅を持って伝える必要があります。
🩺 院長コラム【遺伝カウンセリングで大切にしていること】
16p12.1欠失の遺伝カウンセリングで最も難しいのは、「予後が予測できない」という不確実性をどう伝えるかです。「欠失があるから病気」でも「欠失があっても大丈夫」でもなく、「感受性因子として働くが、発症するかどうかは現時点では予測できない」という事実を正直にお伝えしています。
特に親が同じ欠失を持ちながら健康である場合、それは子どもの予後にとってある程度の安心材料になりますが、完全に同じ経過をたどる保証はありません。「Two-Hitモデル(ツーヒットモデル)」により、子どもには親にはない別の遺伝的要因がある可能性があるからです。
臨床遺伝専門医として、これまでのべ10万人以上のご家族の意思決定と向き合ってきました。不安を抱えている方は、ぜひ一度ご相談ください。
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。16p12.1欠失を含む染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。全染色体検査や微小欠失検査も対応可能です。
🏥 院内で確定検査まで対応
羊水検査・絨毛検査も院内で実施可能。転院の必要がなく、心理的負担を軽減できます。
👩⚕️ 臨床遺伝専門医が常駐
検査前後の遺伝カウンセリングを担当。結果の説明から今後の選択肢まで、専門家が寄り添います。
💰 互助会制度で費用面も安心
互助会(8,000円・NIPT受検者全員に適用)により、陽性時の確定検査(羊水検査)費用が全額補助されます。上限なしで安心です。
よくある質問(FAQ)
🏥 一人で悩まないでください
16p12.1欠失について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- [1] Girirajan S, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42(3):203-209. [PubMed]
- [2] Antonacci F, et al. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat Genet. 2010;42(9):745-750. [PubMed]
- [3] Girirajan S, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367(14):1321-1331. [PubMed]
- [4] Pizzo L, et al. Functional assessment of the “two-hit” model for neurodevelopmental defects in Drosophila and X. laevis. PLoS Genet. 2021;17(4):e1009112. [PubMed]
- [5] Lasser M, et al. 16p12.1 Deletion Orthologs are Expressed in Motile Neural Crest Cells and are Important for Regulating Craniofacial Development in Xenopus laevis. Front Genet. 2022;13:833083. [PubMed]
- [6] Jensen M, et al. Genetic modifiers and ascertainment drive variable expressivity of complex disorders. Cell. 2025. [ScienceDirect]
- [7] OMIM #136570 – Chromosome 16p12.1 Deletion Syndrome, 520-kb. [OMIM]
- [8] GeneReviews – 16p12.2 Recurrent Deletion. [GeneReviews]
- [9] DECIPHER Database – Recurrent 16p12.1 microdeletion. [DECIPHER]
- [10] Unique – 16p12.2 deletions. [Unique]
関連記事






