目次
- 1 1. 12番染色体異常の遺伝子マッピングと主要症候群の局在
- 2 2. 12番染色体トリソミー(完全型およびモザイク型)の病態生理と臨床像
- 3 3. 12番染色体短腕(12p)の部分トリソミーと重複症候群
- 4 4. Pallister-Killian症候群(PKS)との厳密な鑑別
- 5 5. 12番染色体長腕(12q)の部分トリソミーと重複症候群
- 6 6. 12番染色体短腕(12p)の部分モノソミーと間質性欠失症候群
- 7 7. 12番染色体長腕(12q)の部分モノソミーと欠失症候群
- 8 8. 体細胞突然変異としての12番染色体トリソミー:慢性リンパ性白血病(CLL)における意義
- 9 9. 診断アプローチと遺伝カウンセリングの重要性
- 10 よくある質問(FAQ)
- 11 関連記事
- 12 参考文献

📍 クイックナビゲーション
ヒトの12番染色体は、およそ1億3,200万塩基対から構成され、細胞の成長、発生、および機能維持に不可欠な数千の遺伝子を含む極めて重要な遺伝情報のリポジトリ(保管庫)です。
この染色体に構造的、あるいは数的な異常が生じると、影響を受ける遺伝子群の役割に応じて、広範かつ複雑な臨床症状を引き起こします。染色体全体が過剰に存在する「トリソミー」、一部の細胞のみが異常を有する「モザイク型」、染色体の一部が重複する「部分トリソミー(重複)」、そして一部が欠落する「部分モノソミー(欠失)」など、その形態は多岐にわたります。
本記事では、12番染色体に関連するこれらの異常について、最新の分子遺伝学的知見、遺伝子型と表現型(症状)の相関、臨床的な予後、さらには白血病などの腫瘍における体細胞突然変異としての意義に至るまで、臨床遺伝専門医の視点から網羅的かつ詳細に解説します。
1. 12番染色体異常の遺伝子マッピングと主要症候群の局在
染色体は、中央のくびれである「セントロメア(動原体)」を境にして、短い方を「短腕(p)」、長い方を「長腕(q)」と呼びます。12番染色体の異常は、欠失や重複が起きた位置(バンド)によって、引き起こされる症候群が明確に異なります。
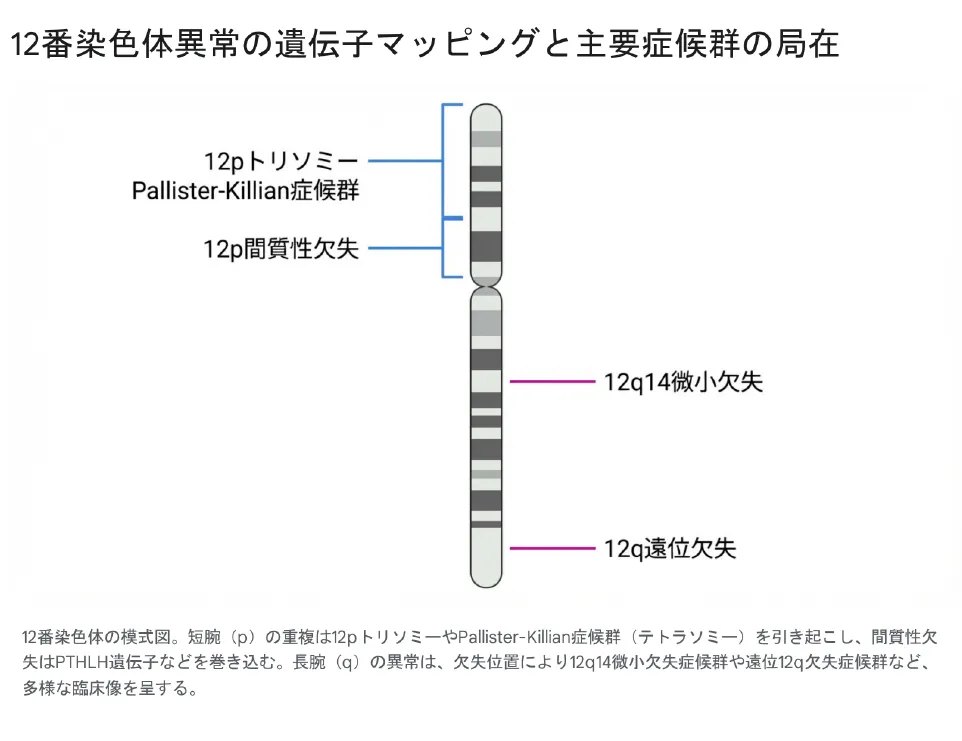
▲ 12番染色体の模式図。短腕(p)の重複は12pトリソミーやPallister-Killian症候群(テトラソミー)を引き起こし、間質性欠失はPTHLH遺伝子などを巻き込みます。長腕(q)の異常は、欠失位置により12q14微小欠失症候群や遠位12q欠失症候群など、多様な臨床像を呈します。
2. 12番染色体トリソミー(完全型およびモザイク型)の病態生理と臨床像
通常、染色体は父親から1本、母親から1本の計2本(二倍体)を受け継ぎます。しかし、細胞分裂のエラーにより12番染色体が3本になってしまう状態を「トリソミー12」と呼びます。
すべての細胞系列において12番染色体が3本存在する「完全型12番染色体トリソミー」は、胚の生存に必須な遺伝子群の極端な過剰発現を引き起こすため、極めて致死性が高い状態です。事実として、完全型トリソミー12の生体例(生きて生まれてきたケース)は医学文献において報告されておらず、通常は妊娠初期に自然流産に至ります。
モザイク型トリソミー12の疫学とメカニズム
しかしながら、受精後の有糸分裂の過程で染色体がうまく分かれなかった結果、正常な細胞(46, XX または 46, XY)と、トリソミーの細胞(47, XX, +12 または 47, XY, +12)が混在する「モザイク型12番染色体トリソミー」となることがあります。この場合、正常な細胞が存在することで致死的な発生異常が部分的に緩和され、妊娠を継続して出生に至る可能性があります。
💡 専門用語解説:モザイク(Mosaicism)
1つの個体の中に、異なる遺伝情報(染色体の数や構造)を持つ細胞がモザイク画のように混ざり合っている状態のこと。モザイクの割合や、異常細胞がどの臓器に多く分布しているかによって、症状の重さが大きく変動します。
モザイク型トリソミー12は極めて稀な疾患と考えられてきましたが、一部の研究では出生500人に1人の割合で予想以上に頻繁に遭遇するというデータも提示されています。羊水検査を実施する際、比較的よく観察される所見の一つです。
NIPTにおける「限局性胎盤モザイク(CPM)」の罠
出生前診断において、非常に重要な概念があります。それが「限局性胎盤モザイク(Confined Placental Mosaicism: CPM)」です。
💡 専門用語解説:限局性胎盤モザイク(CPM)
異常な細胞(トリソミー細胞)が、胎児の体を形作る組織には存在せず、「胎盤」などの胎児外組織にのみ局在している現象のこと。
NIPT(非侵襲的出生前検査)は、母体の血液中に溶け出した「胎盤由来のセルフリーDNA」を分析しています。そのため、このCPMが存在する場合、胎児自身は全くの正常核型であるにもかかわらず、NIPTでは「トリソミー12陽性」という「偽陽性」の結果が出てしまうことがあります。
したがって、NIPTで陽性判定が出た場合には、遺伝カウンセリングと包括的な超音波検査を提供し、羊水検査などの確定的な侵襲的診断を実施することが国際的な学会によって強く推奨されています。
🔗 関連する専門記事
臨床的特徴と多臓器への影響
モザイク型トリソミー12の表現型(症状)は、全くの無症状から、重篤な多臓器奇形に至るまで極端に多様です。主要な症状は以下の多岐にわたるシステムに影響を及ぼします。
- 成長および神経発達: 発達遅滞や成長遅延はほぼ普遍的に見られ、重度の知的障害を伴うことが多いです。低身長も一般的な特徴です。
- 頭蓋顔面異形態: 塔状頭(turricephaly:頭蓋骨の縫合線が早期に癒合し頭が縦に伸びる)、広く高い前額部、眼裂斜下(タレ目)、低位で後方に回転した耳介など、特異な顔貌が形成されます。
- 心血管系異常: 心房中隔欠損症(ASD)や動脈管開存症(PDA)などの先天性心疾患が頻繁に報告されています。心臓の発生はモザイク細胞の存在に極めて敏感です。
- 筋骨格系および皮膚: 筋緊張低下(フロッピーインファント)は早期運動発達を妨げます。皮膚においては、色素性異形成(白斑などのまだら模様)が生じることがあり、これはメラノサイト(色素細胞)の遊走におけるモザイクパターンの直接的な現れと考えられます。
- 感覚器系異常: 感音難聴や網膜症などの異常が報告されています。
予後と個体差の不確実性
特筆すべきは、「血液や皮膚におけるモザイク率(異常細胞の割合)」と「症状の重症度」の間に、予測可能な明確な相関関係が存在しないことです。
例えば、過去の報告例では以下のような極端なばらつきがあります。
- 31歳男性(末梢血モザイク率7%): カルタゲナー症候群(内臓逆位)、慢性副鼻腔炎、不妊症などを呈しつつ成人。
- 36歳女性(皮膚線維芽細胞モザイク率13%): 重度の知的障害、小頭症、広範な白質疾患、低身長、脊柱側弯症など重度な症状。
- 生後5週の女児(皮膚線維芽細胞モザイク率25%): 複雑な先天性心疾患に対する外科手術を試みるも死亡。
羊水検査で出生前にモザイクが検出されても、出生後の血液検査では異常細胞が確認されないケースも散見されます。これは、異常細胞のクローンが脳や心臓など、生検(組織採取)が困難な深部組織にのみ生着している可能性を示唆しており、出生前診断の時点での予後予測を極めて困難にしています。
3. 12番染色体短腕(12p)の部分トリソミーと重複症候群
12番染色体の「短腕(p)」のコピーが通常より1つ多く、合計3つ存在する稀な染色体構造異常です。およそ出生5万人に1人の頻度で発生すると推定されています。
発生機序(なぜ起こるのか?)
大半の症例は、両親のいずれかが保因する「均衡型転座」が、減数分裂(卵子や精子が作られる過程)で不適切に分離することによって生じます。
💡 専門用語解説:均衡型転座(Balanced Translocation)
染色体の一部がちぎれて別の染色体と入れ替わっているが、遺伝物質の「総量」に過不足がない状態。親自身には全く症状がありませんが、子どもに遺伝する際に遺伝子の過不足(不均衡)が生じるリスクが上昇します。
一方で、親からの遺伝ではない「新生突然変異(de novo異常)」として生じるケースも多数報告されています。これは受精前後のDNA配列のミスマッチが原因です。
ダウン症候群との誤診リスクと身体的特徴
新生児の多くは正期産で生まれ、出生体重は正常か、むしろ平均より重い(過体重)傾向にあり、大頭症(相対的に大きな頭部)を伴うことも一般的です。
顔面の特徴としては、長い顔、広く高い前額部、両眼開離、短い眼裂、平坦でふっくらとした頬などが挙げられます。歯科的問題も顕著で、下顎の突出による噛み合わせの不整合や、高い虫歯発生率が報告されており、継続的な専門的ケアが不可欠です。
重要な臨床的課題として、これらの特異な顔貌や、標準的な染色体検査(Gバンド法)での見え方が「21番染色体トリソミー(ダウン症候群)」に酷似している点が挙げられます。そのため初期段階でダウン症と誤診されるケースが多く、正確な診断にはマイクロアレイなどの分子遺伝学的確認が必須です。
神経発達と行動特性の「二極化」
ほぼすべての患者において、重度の知的障害と発達遅滞が認められます。特に言語発達の障害が顕著で、多くの患者は非言語的(non-verbal)であり、ジェスチャーを通じてのみコミュニケーションを図ります。てんかん発作も非常に一般的な合併症で、通常7〜8歳頃に発症する傾向があります。
行動プロファイルには興味深い両極端が見られます。長期フォローアップ研究において、「非常に社交的で明るい」と評される患者がいる一方で、自傷行為、自己刺激行動、気分の激しい波、攻撃性などの重度の行動問題を示す患者も同程度存在することが確認されています。
4. Pallister-Killian症候群(PKS)との厳密な鑑別
12pトリソミーの診断において、絶対に混同してはならないのが「Pallister-Killian症候群(PKS)」です。歴史的にこれらが同一視されることがありましたが、遺伝学的に明確に区別されるべき別個の疾患です。
PKSは、過剰な「同腕染色体(isochromosome 12p)」の存在によって定義されます。
💡 専門用語解説:同腕染色体(Isochromosome)とテトラソミー
染色体が分裂する際のエラーにより、2つの同じ腕(この場合は12番染色体の短腕2つ)がくっついてしまった異常な染色体のこと。結果として、PKSの患者の細胞内には通常の2つの短腕に加え、同腕染色体上の2つの短腕が存在し、合計で短腕が4コピー存在する「テトラソミー12p」の状態となります。
さらにPKSは、正常細胞とテトラソミー細胞が混在するモザイク形態をとることが多く、「血液中からは異常細胞が急速に消失し、皮膚線維芽細胞でのみ確認されやすい」という診断上の厄介な特徴を持ちます。
表現型としては、12pトリソミー(3コピー)とPKS(4コピー)は特異な顔貌などでオーバーラップを示しますが、一般にPKS(テトラソミー)の方がより重篤な症状を呈する傾向にあり、特有の色素性皮膚異常を伴います。
5. 12番染色体長腕(12q)の部分トリソミーと重複症候群
12番染色体短腕(12p)の異常と比較して、長腕(12q)の広範な部分トリソミーに関する臨床報告は極めて稀です。長腕の大部分のコピー数増加は、ゲノムの広範な不均衡をもたらし、胚発生において致命的なエラーを引き起こすことが多いからです。
しかし、稀に生存して出生するケースがあります。家族性の転座に起因する広範な12qトリソミーの症例や、特殊な「挟動原体逆位(pericentric inversion)」に由来する組換え染色体を持つ患者の長期追跡調査が報告されています。
これらの患者は、重度の精神発達遅滞(mental retardation)、難治性のてんかん発作、および複合的な多発奇形を持続的に示しており、12qの過剰な遺伝子量が中枢神経系の成熟プロセスにいかに深刻かつ永続的な障害をもたらすかを浮き彫りにしています。
6. 12番染色体短腕(12p)の部分モノソミーと間質性欠失症候群
12番染色体短腕(12p)の欠失は、コピー数が正常な2つから1つに減少する「モノソミー」状態を引き起こします。特に、染色体の末端ではなく中間の領域が欠落する「間質性欠失」は極めて稀な構造異常です。
表現型の特徴と頻度
データベースの分析により、12p間質性欠失症候群の主要な症状が明らかになっています。
- 頭蓋顔面および眼科的異常(82%): 低位で異形成の耳介、陥没した広い鼻梁、丸い顔、両眼開離など。半数(50%)に視神経萎縮や外斜視などの眼科的問題が合併します。
- 四肢および末端の異常(77%): 骨格系の発生異常を強く反映しており、「短指症(brachydactyly)」が極めて特徴的です。異常に幅の広い親指(母指・母趾)なども確認されます。
- 神経発達・精神医学的特徴: 精神運動遅滞(50%)、知的障害(32%)。発生頻度は9%と比較的低いものの、自閉症スペクトラム障害(ASD)やADHDなどの神経精神医学的特徴を示す患者も存在します。
最小責任領域(MCR)とキーとなる病因遺伝子
近年の高解像度マイクロアレイ染色体検査(CMA)や全エクソームシーケンス(WES)の進歩により、この複雑な症状を決定づける「最小責任領域(Minimal Critical Region: MCR)」が、12p11.23〜12p11.22の約809.94 Kbという極めて狭い範囲に絞り込まれました。
この領域内にある遺伝子が「ハプロ不全」(片方のアレルが失われ、機能が半減して病態を引き起こすこと)に陥ることが病気の核心です。
🧬 骨格形成を司る2つの重要遺伝子
・PTHLH遺伝子: 骨と軟骨の発生メカニズムの中心。欠失すると骨の成熟が早期に止まり、常染色体顕性(優性)の「短指症E2型」の直接的な原因となります。
・RUNX2遺伝子: 骨芽細胞の分化に不可欠なマスター転写因子。欠失すると、鎖骨の欠損や泉門の閉鎖遅延を伴う「鎖骨頭蓋異形成症(CCD)」を引き起こします。
その他にも、中枢神経系で高発現し神経発達障害に関連するDENND5B遺伝子などがこの領域に存在しています。
🔗 関連する最新の検査手法
7. 12番染色体長腕(12q)の部分モノソミーと欠失症候群
12番染色体長腕の欠失(12qモノソミー)は、染色体上のどの部位が欠落したかによって、臨床症状が全く異なる複数の独立した症候群を形成します。大半は生殖細胞形成過程でのランダムなエラーによる新生突然変異(de novo)です。
近位12q欠失(12q11, 12q12, 12q13領域)
セントロメアに近い近位領域の欠失です。出生体重が著しく低く(SGA)、哺乳不良が極めて一般的です。特に12q12の欠失は、クロマチンリモデリングに関与するARID2遺伝子の喪失を伴い、明確な神経発達の遅延、粗な顔立ち、そして約23%に心血管系問題(心房中隔欠損症など)を引き起こします。
12q14微小欠失症候群
12q14領域の微細な遺伝物質の喪失は、常染色体顕性(優性)パターンの遺伝形式をとることがある極めて特異的な症候群を引き起こします。この症候群の中核となる4つの臨床特徴は以下の通りです。
- 軽度の知的障害: 他の欠失領域に比べ、比較的マイルドな認知障害にとどまります。
- 著しい低身長: 骨の長軸方向への成長が強く阻害されます。
- 成長不良と哺乳困難: 出生直後から深刻な哺乳不良が見られ、胃瘻(ガストロストミー)の造設を必要とするケースが多々あります。
- 骨斑紋症(Osteopoikilosis): X線画像上、骨の内部に多数の点状または斑状の硬化像(密度の高い影)が観察される良性の骨形成異常です。
骨斑紋症および低身長の直接的な原因は、欠失領域内部に位置するLEMD3遺伝子の機能喪失であることが分かっています。12q14欠失に関するより専門的な解説は以下のリンクをご参照ください。
🔗 関連する専門記事
遠位12q欠失症候群(Monosomy 12qter)
長腕の末端領域(テロメア付近)の欠失です。小頭症、眉毛癒合(左右の眉毛が中央で繋がる)、大きく丸い団子鼻、異常に小さな耳介など、顕著で多彩な頭蓋顔面異形態をもたらします。発達の全般的な遅延や特有の行動異常をもたらします。
8. 体細胞突然変異としての12番染色体トリソミー:慢性リンパ性白血病(CLL)における意義
これまでに詳述した「生まれつきの異常(生殖細胞系列)」とは全く対照的に、人生の途中で特定の血液細胞にのみ後天的に生じる「体細胞突然変異」としてのトリソミー12も、極めて重要な臨床的意義を持ちます。
最も代表的なのが、欧米の成人白血病の中で最も頻度の高い「慢性リンパ性白血病(CLL)」です。CLL患者の10〜25%において、白血病細胞にのみトリソミー12が検出されます。
- 細胞表面マーカーの特異性: トリソミー12を持つCLL細胞は、CD38、CD49d(インテグリンa4)、CD24などの発現が著しく増強しており、これが腫瘍細胞の組織への浸潤や生存に有利に働くと考えられています。
- ドライバー変異との関連: CLLにおける強力な予後不良因子であるTP53変異は稀ですが、最大40%のトリソミー12陽性患者においてNOTCH1遺伝子の活性化変異が検出され、これが病状の急速な進行を引き起こす主要なドライバーとなっています。
- 予後: 11q欠失や17p欠失を持つ高リスク群よりは良好ですが、13q欠失単独の低リスク群よりは不良である「中間的な予後リスク」に分類されます。
9. 診断アプローチと遺伝カウンセリングの重要性
12番染色体の異常は非常に多様な臨床像を呈するため、精密医療の時代において正確な病態の把握と予後予測には多層的な診断アプローチが要求されます。
従来のGバンド法による染色体検査は全体像を把握するのに有用ですが、12q14微小欠失などの数Mb以下の微細なコピー数変異(CNVs)を検出することは不可能です。そのため、現代の臨床遺伝学においては、FISH法や、ゲノム全体を高解像度でスキャンするマイクロアレイ染色体検査(CMA)、全エクソームシーケンス(WES)の併用が診断のゴールドスタンダードとなっています。
出生前診断と臨床遺伝専門医の役割
近年、非侵襲的出生前検査(NIPT)の普及により、胎盤モザイク(CPM)に起因する偽陽性のトリソミー12や、微小欠失が偶発的に検出されるケースが増加しています。
🔗 出生前検査に関する関連記事
患者様に構造異常が見つかった場合は、ご両親の染色体検査を実施し、それが「偶発的な突然変異(de novo)」なのか、「均衡型転座」に起因するものなのかを特定することが極めて重要です。これにより、次子への再発リスクが全く異なるからです。
検査結果の不確実性と、本稿で詳述したような表現型の広範な多様性を妊婦様やご家族に正確に伝達し、不安を軽減しつつ適切な確定診断(羊水検査など)へと導く「遺伝カウンセリング」の役割は、今後ますます重要性を増していくと考えられます。

よくある質問(FAQ)
関連記事
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- 12q deletions FTNW – RareChromo.org
- General information about positive NIPT results: Trisomy 12 – Sonic Genetics
- Mosaic trisomy 12 diagnosed in a female patient: clinical features, genetic analysis, and review of the literature – PMC
- Mosaic trisomy 12 syndrome – Orphanet
- Trisomy 12p – Chromosome Disorder Outreach, Inc
- Duplications of 12p – RareChromo.org
- Pallister-Killian syndrome – RareChromo.org
- Partial trisomy 12q: a clinically recognisable syndrome. Genetic risks associated with translocations of chromosome 12q – PMC
- RUNX2 gene: MedlinePlus Genetics
- Chromosome 12q12 Deletion – Chromosome Disorder Outreach, Inc
- 12q14 microdeletion syndrome – Orphanet
- Morphological, immunophenotypic, and genetic features of chronic lymphocytic leukemia with trisomy 12: a comprehensive review – PMC






