目次

ジュベール症候群8型(JBTS8)は、ARL13B遺伝子の変異によって引き起こされる、出生約8〜10万人に1人という極めて希少な神経発達障害です。MRI検査で確認される「大臼歯様サイン(Molar Tooth Sign: MTS)」を特徴とし、小脳・脳幹の発達異常だけでなく、全身の一次繊毛(primary cilium)の機能不全が原因となる「繊毛病(Ciliopathy)」のひとつです。初期には純粋な神経疾患とみなされていましたが、現在では網膜ジストロフィーや肥満リスクを含む多臓器横断的な疾患として理解されています。
Q. ジュベール症候群8型(JBTS8)とはどのような疾患ですか?
A. ARL13B遺伝子の変異によって引き起こされる、極めて希少な常染色体潜性遺伝(劣性遺伝)の繊毛病です。MRI上の大臼歯様サイン(MTS)が診断の鍵となり、神経症状に加えて網膜ジストロフィーや肥満リスクも持つ多臓器疾患です。日本では指定難病177として医療費助成の対象となっています。
- ➤疾患の定義 → OMIM 612291、指定難病177、出生約8〜10万人に1人、JS全体の2%未満
- ➤分子メカニズム → ARL13BのGEF機能喪失→繊毛内タンパク質輸送の空間的制御が破綻
- ➤主な症状 → 筋緊張低下・小脳性運動失調・発達遅滞、網膜ジストロフィー、肥満リスク
- ➤鑑別診断 → 有馬症候群(CEP290型)・AHI1型・TMEM67型との違いを解説
- ➤診断・管理 → MRI所見+遺伝子検査、臓器別の生涯サーベイランスプロトコル
1. ジュベール症候群8型(JBTS8)とは:疾患の定義と歴史的背景
ジュベール症候群(Joubert syndrome: JS)は、1969年にフランスのMarie Joubertによって初めて記載された希少な神経発達障害です。現在40種類以上の原因遺伝子が同定されており、それぞれ「JBTS1、JBTS2……」とサブタイプに分類されています。そのうち、第3染色体長腕(3q11.1-q11.2)に位置するARL13B遺伝子の変異が原因となるサブタイプが「ジュベール症候群8型(JBTS8)」です(OMIM: 612291)。
世界的な有病率はJS全体で出生80,000〜100,000人に1人と推定されており、その中でARL13B変異が占める割合は2%未満と特に稀です。軽症例や未診断例も多く存在するため、実際の罹患者数はさらに多い可能性があります。
💡 用語解説:常染色体潜性遺伝(劣性遺伝)とは
「常染色体」とは、性染色体(X・Y)以外の染色体のこと。「潜性(劣性)」とは、2本の染色体の両方に変異が揃ったときに症状が現れる遺伝形式です。つまり、親がそれぞれ1つずつ変異を持っていても(保因者)症状は現れません。両親が保因者の場合、子どもに疾患が発症する確率は理論上25%です。
JSの放射線学的な最大の特徴は、頭部MRI検査の横断像(体軸断)で確認できる「大臼歯様サイン(Molar Tooth Sign: MTS)」です。これは、①小脳虫部の低形成または無形成、②異常に深く変形した脚間窩、③水平方向に肥厚・延長した上小脳脚——という3つの解剖学的異常が組み合わさって現れる特異な画像所見で、JSを診断するための必須要件です。
💡 用語解説:大臼歯様サイン(Molar Tooth Sign: MTS)
MRI画像で脳幹と小脳の接合部を横断したときに、その形が「奥歯(大臼歯)」に似て見えることから名付けられた所見です。小脳虫部(左右の小脳をつなぐ部分)が正常に発達していないこと、脳幹の特定部位の形が変形していることによって生じます。ジュベール症候群の確定診断に必須の所見であり、これが認められるかどうかが診断の出発点となります。
歴史的にJSは中枢神経系だけの疾患と考えられてきましたが、21世紀以降の分子遺伝学の進歩により、JSの原因遺伝子産物のほぼすべてが「一次繊毛(Primary cilium)」またはその基底小体に局在することが判明しました。これによりJSは、全身の臓器に存在する一次繊毛の機能不全に起因する「繊毛病(Ciliopathy)」のスペクトラムの一つとして再定義されています。
💡 用語解説:繊毛病(Ciliopathy)とは
体のほぼすべての細胞に存在する微細な突起構造「一次繊毛(primary cilium)」の形成・機能の異常によって引き起こされる疾患群の総称です。一次繊毛は細胞のアンテナのように、外部からのシグナルを感知して細胞内に伝達する重要な役割を担っています。繊毛が正常に機能しないと、脳・眼・腎臓・肝臓など多くの臓器に同時に異常が生じます。ジュベール症候群以外にも、バルデ・ビードル症候群、腎ネフロン癆、メッケル・グルーバー症候群なども繊毛病に含まれます。
🇯🇵 日本の制度的位置づけ:ジュベール症候群関連疾患(JSRD)は厚生労働省の「指定難病177」に認定されており、診断が確定した患者と家族は医療費助成制度の対象となります。JBTS8はJSRDの中の「有馬症候群以外のジュベール症候群関連疾患」カテゴリーに分類されます。
2. 原因遺伝子ARL13Bと分子病態メカニズム
JBTS8の原因遺伝子であるARL13B(ADP-ribosylation factor-like GTPase 13B)は、Rasスーパーファミリーに属する低分子量GTPアーゼをコードする遺伝子です。ARL13Bタンパク質は脊椎動物の一次繊毛の形成に必須であり、繊毛内に極めて高い濃度で局在しています。この局在は、タンパク質のパルミトイル化(脂質修飾)によって厳密に制御されています。
💡 用語解説:ARL13B遺伝子とは
ARL13Bは「低分子量GTPアーゼ」と呼ばれるタンパク質の一種をコードする遺伝子です。GTPアーゼはGTP(細胞のエネルギー通貨の一種)に結合することで「オン」になり、GDPに変換されることで「オフ」になる分子スイッチとして機能します。ARL13Bは一次繊毛の中に特に高濃度で存在し、繊毛内のタンパク質輸送を制御するという重要な役割を担っています。OMIMデータベースでは遺伝子座番号608922として登録されています。
2.1 GEFとしての役割:繊毛内タンパク質輸送の空間的制御
ARL13Bの最も重要な機能は、別の繊毛関連GTPアーゼであるARL3に対するGEF(グアニンヌクレオチド交換因子)として働くことです。ARL13Bは一次繊毛内に限定的に局在しているため、ARL3の活性化が「繊毛の中だけ」で起こるという空間的な制御が実現しています。
💡 用語解説:GEF(グアニンヌクレオチド交換因子)とは
GEF(Guanine nucleotide Exchange Factor)は、GTPアーゼを不活性型(GDP結合型)から活性型(GTP結合型)に変換する「触媒タンパク質」です。ARL13BはGEFとして働き、ARL3というタンパク質を活性化します。活性化されたARL3は、視覚に必須の「PDE6」など重要なタンパク質を繊毛内の正確な場所へ届けます。JBTS8患者で確認されているR79QやR200Cなどの変異は、このGEF活性を著しく低下させます。
活性化されたARL3(ARL3-GTP)は、PDE6δやUNC119a/bといったキャリアタンパク質に結合した脂質修飾カルゴ(輸送標的タンパク質)を、繊毛内において正確に解離・放出させます。この「繊毛内限定の活性化」という空間的制御機構が破綻すると、繊毛内への必須タンパク質の供給不足が生じ、多臓器不全の原因となります。
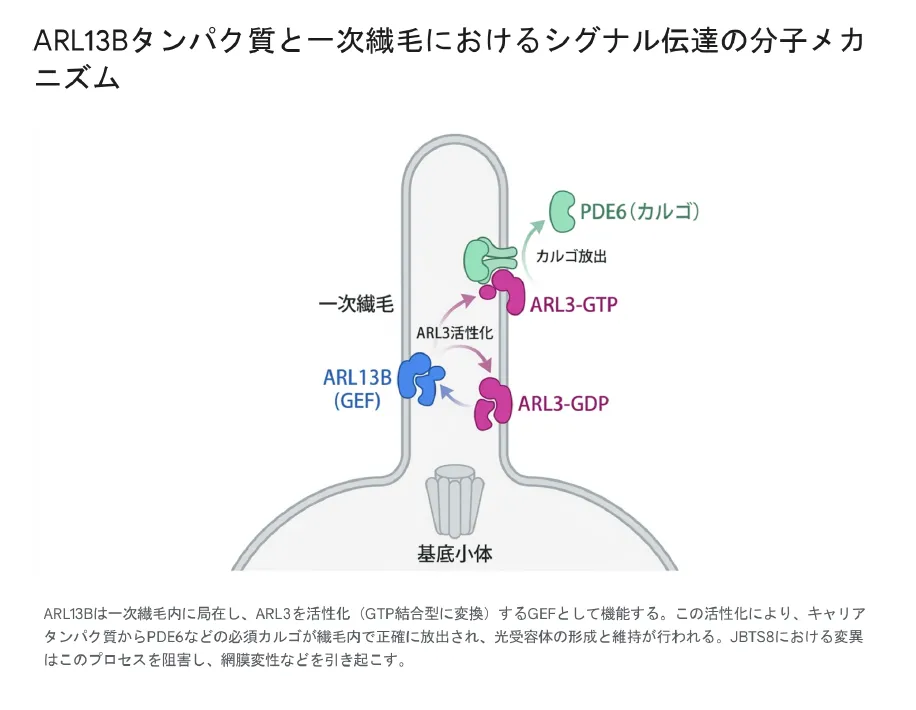
ARL13Bは一次繊毛内に局在し、ARL3をGTP結合型(活性型)に変換するGEFとして機能する。活性化されたARL3はキャリアタンパク質からPDE6などの必須カルゴを繊毛内で正確に放出させる。JBTS8における変異はこのプロセスを阻害し、網膜変性などの多臓器症状を引き起こす。
2.2 エキソシスト複合体との協調:腎臓・組織恒常性への関与
ARL13Bの分子機能はGEF活性に留まりません。細胞内小胞輸送を制御するエキソシスト複合体のサブユニット(Sec8・Sec5・Exo70)と直接結合することが示されています。ゼブラフィッシュを用いた研究では、ARL13BまたはSec10の欠失が腎臓の発達異常や全身の水腫を引き起こすことが確認されており、ARL13Bとエキソシスト複合体が同一の経路で協調して一次繊毛の構築と腎臓などの組織恒常性の維持に寄与していることが証明されています。
2.3 ソニック・ヘッジホッグ(Shh)シグナルと脳・小脳の発達
胎児の発達段階において、一次繊毛はソニック・ヘッジホッグ(Sonic Hedgehog: Shh)シグナル伝達のハブとして機能します。ARL13BはこのShhシグナル経路の適切な活性化に不可欠です。ARL13B欠失モデルでは、Shhシグナルの減弱により小脳顆粒細胞の増殖が阻害され、小脳虫部・脳幹の正常な発達が妨げられます。これがMRI上のMTSを形成する根本的なメカニズムです。
💡 用語解説:ハイポモルフ変異とは
タンパク質の機能を完全に失わせるのではなく、部分的に低下させる変異のことです。動物モデルでのARL13B完全欠失は神経管閉鎖障害など致死的な重症表現型を示しますが、ヒトのJBTS8患者の多くは出生後に生存できます。これはヒトで同定されているARL13B変異の多くがハイポモルフ変異であるためと考えられています。機能低下に対する閾値の差が、臓器ごとの症状の現れ方の違いをもたらしています。

3. 主な症状と臨床的スペクトラム
JBTS8の臨床像は、当初は「純粋な神経疾患」と考えられていましたが、全エクソーム解析などの普及により、網膜ジストロフィーや肥満を含む多臓器横断的な表現型を示すことが明らかになっています。同じARL13B変異でも、症状の現れ方や重症度は患者によって異なります。
🧠 神経・発達・形態
- 新生児期の重度な筋緊張低下(Hypotonia)
- 成長後に移行する小脳性運動失調(Ataxia)
- 軽度〜重度の知的発達遅滞(ほぼ全例)
- 自閉症スペクトラム様の行動・情緒不安定
- 乳児期の不規則な呼吸パターン(無呼吸・多呼吸)
- てんかん発作(約10%)
- 巨頭症・突出した前頭部・眼窩開離などの特徴的顔貌
👁️ 視覚・眼科(JBTS8の特徴)
- 網膜ジストロフィー(進行性の視細胞変性)
- 眼球運動失行(OMA)・眼振・斜視
- 光受容体の構造異常(外節形成不全)
- 視覚応答の消失(ERG異常)
- 重篤な網膜障害はARL13B変異の重要な臨床指標
⚖️ 代謝・肥満(新たに判明)
- 病的肥満(視床下部繊毛機能不全が原因)
- 満腹シグナルの受容障害による食欲制御困難
- JSでは歴史的に稀とされてきたが、ARL13B変異では報告あり
- 網膜障害と肥満の併発例でARL13B新規ミスセンス変異を同定
🫘 腎臓・肝臓
- JSRD全体では腎疾患23〜38%、先天性肝線維症15%
- JBTS8での重篤な腎・肝不全は他の変異型より相対的に低頻度
- ただし家族間差異があるため生涯監視が必要
- 動物モデルではエキソシスト機能不全による腎嚢胞形成を確認
💡 用語解説:網膜ジストロフィーとは
網膜(眼の奥にある光を感じる膜)の視細胞が徐々に変性・消失していく病気の総称です。ARL13BのGEF機能が失われると、視覚に必須のタンパク質(PDE6など)が光受容体の外節へと正常に輸送されなくなります。その結果、光受容体円板の層状構造が形成されず、視覚応答が失われます。乳幼児期から徐々に視力低下が進行し、放置すれば重篤な視機能障害に至ります。定期的な網膜電図(ERG)検査による早期発見が重要です。
視床下部繊毛機能不全と肥満:JBTS8における近年の重要な発見
JBTS8の臨床スペクトラムにおける近年の最も革新的な発見は、「病的肥満との関連」です。Thomasらの研究(2015年)では、網膜障害と肥満を併発したJBTS8患者においてARL13Bの新規ミスセンス変異が同定され、ARL13Bタンパク質が新生児マウスの視床下部ニューロンの一次繊毛内に極めて高い濃度で局在していることが突き止められました。視床下部は全身のエネルギー代謝と食欲調節を司る中枢であり、この部位における繊毛機能の喪失が満腹シグナルの受容障害を引き起こし、病的肥満を誘発すると考えられています。
4. 鑑別診断:他のジュベール症候群サブタイプとの違い
JSRDは40種類以上の原因遺伝子によるサブタイプが存在し、いずれも大臼歯様サインを共有します。遺伝子ごとに合併しやすい臓器病変が異なるため、原因遺伝子の特定は管理方針の個別化に直結します。下表はJBTS8(ARL13B)と主な比較対象を示しています。
| 遺伝子(サブタイプ) | JS全体の割合 | 特徴的な合併症 |
|---|---|---|
| ARL13B(JBTS8) | <2% | 網膜ジストロフィー、肥満リスク、腎・肝障害は相対的に低頻度 |
| AHI1(JBTS3) | 5〜10% | 高頻度の網膜ジストロフィー |
| CEP290(JBTS5) | 約10% | 早期腎不全・重度網膜変性(有馬症候群)、最重症型 |
| TMEM67(JBTS6) | 5〜10% | 高頻度の先天性肝線維症(COACH症候群) |
💡 用語解説:有馬症候群とは
JSRDの中で特に重症のサブタイプで、乳児期早期からの進行性腎機能障害(ネフロン癆)と重度の網膜ジストロフィーを主な特徴とします。主にCEP290遺伝子の変異が原因で、小児期から腎透析が必要となることがあります。日本の指定難病の診断基準では有馬症候群とそれ以外のJSRDを明確に区別しており、JBTS8は「それ以外のJSRD」に分類されます。
💡 用語解説:ネフロン癆(ネフロンロウ)とは
腎臓の機能単位「ネフロン」が徐々に破壊されていく遺伝性の腎疾患です。多尿・多飲・夜尿などの症状から始まり、進行すると腎不全に至ります。JSRDの約23〜38%に合併するとされますが、ARL13B変異(JBTS8)での頻度はCEP290変異に比べて相対的に低いとされています。ただし、長期的なモニタリングは欠かせません。
5. 診断基準と遺伝子検査の進め方
5.1 日本の診断基準(指定難病177の枠組み)
日本において、JBTS8を含む「有馬症候群以外のJSRD」の確定診断(Definite)には、以下の要件を組み合わせた判定が行われます。
📋 JBTS8確定診断に必要な主要要件
- ➤主要症状A1:精神運動発達遅滞(知的障害・発達遅滞)
- ➤主要症状A2:筋緊張低下または運動失調
- ➤画像所見B1:頭部MRIにおける大臼歯様サイン(MTS)の確認
- ➤遺伝学的検査D:ARL13B遺伝子における病的変異(両アレル)の同定
医療費助成の対象となる「重症度分類」では、診断の確定に加えて以下のいずれかを満たす必要があります。
- ➤機能障害:modified Rankin Scale(mRS)・食事栄養評価スケール・呼吸評価スケールのいずれかが「3以上」
- ➤腎障害:慢性腎臓病(CKD)重症度分類で末期腎不全リスクの高い「赤」領域
- ➤視覚障害:良好な方の眼の矯正視力が0.3未満
JBTS8患者は、運動失調や重度の発達遅滞によって機能障害スケールを満たすことが多く、またARL13B変異特有のリスクである網膜ジストロフィーが進行した場合には視覚障害の基準からも助成対象となります。
5.2 分子遺伝学的検査:パネル検査から全エクソーム解析まで
💡 用語解説:遺伝子パネル検査・全エクソーム解析とは
遺伝子パネル検査は、関連する複数の遺伝子(数十〜数百種類)を同時に調べる次世代シーケンス(NGS)検査です。JSRD関連の遺伝子が複数含まれるパネルを用いることで、ARL13Bを含む原因遺伝子を効率よく同定できます。全エクソーム解析(WES)は、遺伝子のタンパク質をコードする領域(エクソン)すべてを網羅的に解析する手法で、パネルに含まれない遺伝子の変異も検出できます。患者本人と両親を同時に解析する「トリオ解析」は、変異の由来(どちらの親から遺伝したか)を明確にし、診断精度を高めます。
JSRDに関連する運動失調のNGSパネル検査は、ARL13BをはじめとするJSRD原因遺伝子を効率的にスクリーニングするための有用な出発点となります。遺伝子型と表現型の相関(Gene-phenotype correlations)に基づき、ARL13B変異患者では網膜と体重管理に優先的に注力したサーベイランスプランを立案することが可能になります。
6. 治療・長期管理と多職種連携
JBTS8に対する根本的な治療法(遺伝子治療など)は現在確立されておらず、医療介入の主体は症状の緩和・合併症の予防・生活の質(QOL)の最大化を目的とした対症療法と多職種連携です。遺伝子型の特定により、臓器ごとのリスクに合わせた個別化サーベイランスが可能になります。
6.1 新生児・乳児期:最優先の呼吸・栄養管理
新生児・乳児期に最も緊急を要するのは呼吸管理と栄養摂取の維持です。間欠的な多呼吸と睡眠時無呼吸は、未治療の場合に重篤な低酸素症や肺高血圧症を引き起こすリスクがあります。睡眠医学の専門家によるモニタリングが必要であり、重症例ではカフェインなどの呼吸刺激薬の投与や酸素投与が行われます。嚥下障害が強い場合は誤嚥性肺炎・発育不全(Failure to thrive)を回避するため、胃瘻造設(Gastrostomy tube placement)による経管栄養の導入が推奨されます。
6.2 神経発達的介入:早期リハビリテーションの重要性
早期からの理学療法(PT)・作業療法(OT)・言語聴覚療法(ST)の介入が極めて重要です。運動失調や構音障害に対する代償機能の獲得に専門的なリハビリテーションは不可欠です。臨床報告では、1年間の集中的なリハビリテーションとフォローアップにより患者の能力が劇的に改善したケースが記録されており、早期介入の有効性が裏付けられています。てんかん発作(約10%)に対しては、脳波検査に基づく抗てんかん薬によるコントロールが行われます。
6.3 臓器別のサーベイランス体制
👁️ 眼科・網膜サーベイランス
ARL13B変異でリスクが高まる網膜ジストロフィーに対し、小児眼科医による定期的な網膜電図(ERG)・眼底検査が不可欠。視力低下が進行する場合は早期からロービジョンケアを導入し、学習環境・日常生活の安全を確保する環境調整を行う。斜視・重度の眼瞼下垂には外科的修復術を検討。
⚖️ 代謝・体重管理
視床下部繊毛機能不全による食欲制御困難が予想されるため、定期的な成長曲線の確認と栄養管理が不可欠。早期からの食事介入・行動療法を開始し、思春期以降は内分泌機能評価(ホルモン異常の有無)を実施。
🫘 腎臓・肝臓モニタリング
多尿・多飲などのネフロン癆初期症状を毎年臨床評価。血液検査(BUN・クレアチニン・シスタチンC)を1〜2年ごとに実施。肝臓は腹部超音波検査で門脈圧亢進の兆候(脾腫など)を確認し、血小板数・肝酵素(ALT・GGT)を採血評価。
7. 遺伝カウンセリングの意義
JBTS8の確定診断後、患者と家族への丁寧な遺伝カウンセリングが必要です。常染色体潜性遺伝(劣性遺伝)形式をとることから、家族計画に関する複数の重要事項を専門医とともに整理することが求められます。
- ➤遺伝形式と再発リスク:両親がそれぞれ1つずつ変異を保因している場合(保因者)、次子への遺伝確率は理論上25%です。両親の変異確認(保因者診断)が推奨されます。
- ➤保因者スクリーニング:患者の兄弟姉妹が保因者である可能性があります。将来の家族計画に備えた保因者スクリーニングについて専門医にご相談ください。
- ➤出生前診断の選択肢:次子を望む場合、絨毛検査・羊水検査による出生前遺伝子診断が選択肢として存在します。既知の変異が同定されている場合は確実な診断が可能です。
- ➤長期的な情報収集:JBTS8は症例数が少なく自然歴に関するデータが限られています。医療機関との連携を継続し、新たな知見が得られた際に適宜情報を更新することが重要です。
8. よくある誤解
誤解①「JSは脳だけの病気」
JSはMRI所見(大臼歯様サイン)に代表される脳形態異常が有名ですが、繊毛病として全身の臓器に影響します。特にARL13B変異では網膜ジストロフィーや肥満リスクも管理の対象です。
誤解②「JBTS8は軽症型」
腎・肝障害が相対的に低頻度であることを「軽症」と誤解することがありますが、進行性の網膜変性による視力喪失や病的肥満など、生涯にわたる医療管理を必要とする重篤な合併症があります。
誤解③「両親が健康なら遺伝ではない」
常染色体潜性遺伝では、両親が保因者であれば症状なく変異を持ちます。「両親が健康=遺伝性疾患ではない」という誤解が診断を遅らせることがあります。保因者診断が重要です。
誤解④「MRIでMTSがなければJSではない」
MTSはJSの必須所見ですが、MRI撮影の断面や時期によっては典型的なMTSとして描出されない場合もあります。臨床症状と遺伝子検査を組み合わせた総合的な判断が欠かせません。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少遺伝性疾患の診断・遺伝カウンセリングについて
ジュベール症候群8型をはじめとする繊毛病・希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] Parisi MA. Joubert Syndrome. In: Adam MP, et al., eds. GeneReviews®. Seattle (WA): University of Washington; 1993-2024. [NCBI Bookshelf NBK1325]
- [2] Cantagrel V, et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83(2):170-179. [PMC2495072]
- [3] Thomas S, et al. Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. Eur J Hum Genet. 2015;23(8):1133-1137. [PMC4402632]
- [4] Caspary T, et al. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007;12(5):767-778. Arl13b/scorpion: [PMC2778746]
- [5] Gotthardt K, et al. A G-protein activation cascade from Arl13B to Arl3 and implications for ciliary targeting of lipidated proteins. eLife. 2015;4:e11859. [eLife e11859]
- [6] Seixas C, et al. Arl13b and the exocyst interact synergistically in ciliogenesis. Mol Biol Cell. 2016;27(24):3958-3971. [Mol Biol Cell]
- [7] Dilan TL, et al. ARL13B, a Joubert Syndrome-Associated Protein, Is Critical for Retinogenesis and Elaboration of Mouse Photoreceptor Outer Segments. J Neurosci. 2019;39(8):1347-1364. [J Neurosci]
- [8] 難病情報センター. ジュベール症候群関連疾患(指定難病177). 厚生労働省. [難病情報センター]
- [9] Lu H, et al. Clinical and genetic characteristics of 36 children with Joubert syndrome. Front Pediatr. 2023;11:1102639. [Frontiers in Pediatrics]
- [10] Doherty D. Joubert Syndrome: Insights Into Brain Development, Cilium Biology, and Complex Disease. Semin Pediatr Neurol. 2009;16(3):143-154. [University of Washington]
- [11] Orphanet. Joubert syndrome 8. ORPHA: related ARL13B. [Orphanet ARL13B]






