疾患の別名
疾患概要
ジュベール症候群は、複数の身体部位に影響を及ぼす遺伝性の疾患です。この症候群は患者によって症状が異なり、家族内でもバラエティに富んでいます。
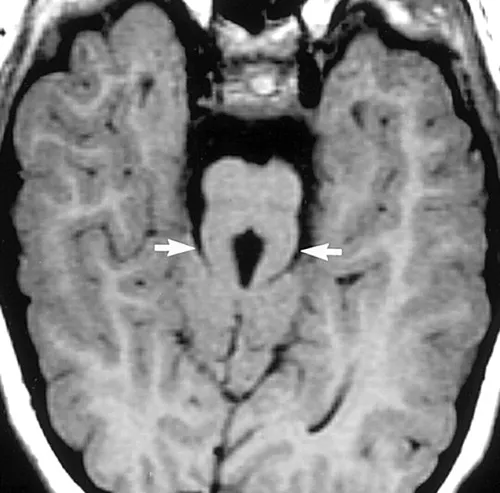
主な特徴は「臼歯徴候」と呼ばれる、脳の異常を示す特徴です。この異常は、脳の後方部分にある特定の構造の異常発達に起因し、MRIで確認できます。この異常はMRIで見ると臼歯の形状に似ており、その名前の由来です。
※臼歯徴候(Molar Tooth Sign)は、ジュベール症候群(Joubert syndrome)など一部の神経疾患で見られる特徴的な脳の異常の一つです。この徴候は、脳のMRI画像において、特定の構造が臼歯の断面に似ていることを指します。
臼歯徴候は、主に脳の後方部分、具体的には小脳海綿体や脳幹などの構造の異常発達に起因します。この異常はMRIで視覚的に確認でき、臼歯の形状に似ていることからこの名前がつけられました。MRIで中脳と橋の境界部(峡部)を通る断面において見ます。
乳幼児期に、筋肉の緊張が低下することが多く、これが幼児期に運動の協調性を乱す原因となります。その他の特徴には、呼吸異常、眼球運動の問題、発達の遅れ、知的障害、特徴的な顔の特徴が含まれます。また、視力の低下を引き起こす網膜ジストロフィーや、眼の構造に問題を引き起こすコロボーマなど、他の目の異常や腎臓疾患、肝臓疾患、骨格の異常、ホルモンの問題など、さまざまな症状が現れることがあります。
以前はこれらの症状が異なる疾患として扱われていましたが、現在では臼歯徴候を伴う場合、すべてジュベール症候群として分類されています。
遺伝的不均一性
JBTS1(213300):INPP5E遺伝子(613037)の変異に起因
JBTS2(608091):TMEM216遺伝子(613277)の変異に起因
JBTS3(608629):AHI1遺伝子(608894)の変異に起因
JBTS4(609583):NPHP1遺伝子(607100)の変異に起因
JBTS5(610188):CEP290遺伝子(NPHP6(610142)とも呼ばれる)の変異に起因
JBTS6(610688):TMEM67遺伝子(609884)の変異に起因
JBTS7(611560):RPGRIP1L遺伝子(610937)の変異に起因
JBTS8(612291):ARL13B(608922)の変異に起因
JBTS9(612285):CC2D2A遺伝子(612013)の変異に起因
JBTS10(300804):CXORF5遺伝子(300170)の変異に起因
JBTS11(613820):TTC21B遺伝子(612014)の変異に起因
JBTS12(200990):KIF7遺伝子(611254)の変異に起因
JBTS13(614173):TCTN1遺伝子(609863)の変異に起因
JBTS14(614424):TMEM237遺伝子(614423)の変異に起因
JBTS15(614464):CEP41遺伝子(610523)の変異に起因
JBTS16(614465):TMEM138遺伝子(614459)の変異に起因
JBTS17(614615):CPLANE1遺伝子(614571)の変異に起因
JBTS18(614815):TCTN3遺伝子(613847)の変異に起因
JBTS19(614844):ZNF423遺伝子(604577)の変異に起因
JBTS20(614970):TMEM231遺伝子(614949)の変異に起因
JBTS21(615636):CSPP1遺伝子(611654)の変異に起因
JBTS22(615665):PDE6D遺伝子(602676)の変異に起因
JBTS23(616490):KIAA0586遺伝子(610178)の変異に起因
JBTS24(616654):TCTN2遺伝子(613846)の変異に起因
JBTS25(616781):CEP104遺伝子(616690)の変異に起因
JBTS26(616784):KATNIP遺伝子(616650)の変異に起因
JBTS27(617120):B9D1遺伝子(614144)の変異に起因
JBTS28(617121):MKS1遺伝子(609883)の変異に起因
JBTS29(617562):TMEM107遺伝子(616183)の変異に起因
JBTS30(617622):ARMC9遺伝子(617612)の変異に起因
JBTS31(617761):CEP120遺伝子(613446)の変異に起因
JBTS32(617757):SUFU遺伝子(607035)の変異に起因
JBTS33(617767):PIBF1遺伝子(607532)の変異に起因
JBTS34(614175):B9D2遺伝子(611951)の変異に起因
JBTS35(618161):ARL3遺伝子(604695)の変異に起因
JBTS36(618413):FAM149B1遺伝子(618763)の変異に起因
JBTS37(619185):TOGARAM1遺伝子(617618)の変異に起因
JBTS38(619476):KIAA0753遺伝子(617112)の変異に起因
JBTS39(619562):TMEM218遺伝子(619285)の変異に起因
JBTS40(619582):IFT74遺伝子(608040)の変異に起因
臨床的特徴
ジュベール症候群の臨床的特徴について、以下の情報が示されています。
Lagier-Tourenneら(2004)による報告では、トルコ系とスイス系の2つのジュベール症候群の家族について述べられています。トルコ家系には5人、スイス家系には2人の患者がいました。これらの患者は全て、早期に筋緊張低下、臼歯徴候、小脳椎体低形成などの特徴的な症状を示しました。また、認知障害、新生児呼吸障害、小脳失調症、眼振、網膜ジストロフィー、視力低下、脊柱後弯症、骨格発育遅延など、さまざまな臨床的特徴が報告されています。
Valenteら(2005)のレビューでは、JBTS3の患者において、多発性小脳症、脳梁奇形、痙攣、痙縮など、中枢神経系に関連する多くの異常が頻繁に見られることが明らかにされています。一方で、腎疾患、肝疾患、多指症などは報告されていないことが示されています。
Utschら(2006)による報告では、JBTS3患者の例として、両親から生まれた2人のパキスタン人兄弟が述べられています。これらの兄弟は、小脳失調、発達遅延、眼振、眼球運動失行などの症状を示しました。また、1人の兄弟はネフローゼにより16歳までに末期腎不全を発症しました。分子生物学的解析により、AHI1遺伝子のホモ接合体変異(608894.0007)が同定されました。この結果から、JBTS3患者では腎病変が発生する可能性が示唆されています。
これらの情報は、ジュベール症候群の臨床的特徴の多様性を示し、異なる遺伝子変異によってさまざまな症状が引き起こされることを示唆しています。
マッピング
また、遺伝子型-表現型研究からは、CORS2遺伝子(608091)とは異なり、JBTS3は腎機能障害とは関連しないことが明らかにされました。つまり、JBTS3の原因遺伝子は腎機能障害には影響を与えない可能性が示されました。
遺伝
しかし、ジュベール症候群の中には「X連鎖劣性遺伝」という稀なケースもあります。X連鎖劣性遺伝の場合、原因となる遺伝子はX染色体(性染色体の一つ)に位置しています。男性はX染色体を1本しか持たないため、このX染色体上の遺伝子が変異していれば病気が発症します。一方、女性はX染色体を2本持っており、病気が発症するためには両方のX染色体上の遺伝子に変異が必要です。女性が両方のX染色体上でこの遺伝子の変異を持つことは非常に珍しいため、男性の方がこのタイプの遺伝子変異による病気を発症する確率が高くなります。
X連鎖遺伝の特徴として、父親は自分のX染色体を息子に遺伝させることができません(男性の子どもは父親からY染色体を受け継ぎます)。そのため、父親から息子へのこの病気の遺伝は起こりません。
ジュベール症候群3は常染色体劣性遺伝です。
頻度
ジュベール症候群を引き起こす特定の遺伝子変異は、アシュケナージ・ユダヤ人、フランス系カナダ人、ハッタイトといった特定の民族グループでより頻繁に見られることが報告されています。これは、遺伝的な背景が疾患の発生に重要な役割を果たしていることを示唆しています。ジュベール症候群に関連する遺伝子変異は、これらの集団の中でより一般的な遺伝的多様性の一部と考えられています。
ジュベール症候群の特徴には、低筋緊張、調整障害、異常な呼吸パターン(特に新生児期)、眼の動きの問題、認知障害などが含まれます。また、MRI検査により特定の脳の異常(「モルヒオンサイン」として知られる)が確認されることが一般的です。これらの症状の幅広いスペクトルが、診断を困難にしている一因です。
遺伝子検査と詳細な臨床評価が、ジュベール症候群の診断において重要です。早期の正確な診断と管理は、これらの患者の予後にとって非常に重要です。
原因
ジュベール症候群に関連する遺伝子に変異がある場合、一次繊毛の構造や機能に問題が生じます。これにより、発達に重要な化学シグナルの伝達経路が妨げられる可能性があります。研究者たちは、一次繊毛の不具合がこの病気の多くの特徴に関係していると考えていますが、どのようにして具体的な発達異常を引き起こすかはまだ完全にはわかっていません。
ジュベール症候群の患者の約60~90%では、既知の遺伝子変異が原因ですが、残りの症例では遺伝的原因はまだ解明されていません。
ジュベール症候群3はAHI1遺伝子の変異に起因します。
分子遺伝学
Ferlandら(2004)の研究では、ジュベール症候群に関連する遺伝子座が染色体6q23.2-q23.3上に同定され、AHI1(Abelson helper integration site-1)遺伝子に3つの致死的変異が発見されました。AHI1遺伝子は特に脳のニューロンで高発現しており、ヒトの進化において重要な役割を果たしていた可能性が示唆されました。
Dixon-Salazarら(2004)の研究では、ジュベール症候群の血縁家系において、AHI1遺伝子に1つのミスセンス変異と2つのフレームシフト変異が同定されました。
Parisiら(2006)の研究では、117人のジュベール症候群のプロバンドをスクリーニングし、AHI1遺伝子の変異について調査しました。その結果、新規変異と既知の変異が同定され、特に網膜ジストロフィーを合併した症例が報告されました。
Valenteら(2006)は、10家系のジュベール症候群患者11例において、AHI1遺伝子の多くの変異を同定し、疾患の原因として特定しました。この研究では、AHI1変異と症状の関連についても詳しく調査されました。
Najmabadiら(2011)の研究では、ジュベール症候群の症候群型および非症候群型を分離するために、ホモ接合体マッピングとエクソン濃縮と次世代シーケンシングを組み合わせて用いました。ジュベール症候群-3の2家族の罹患者において、AHI1遺伝子の変異が同定されました。
これらの研究は、ジュベール症候群の遺伝的基盤を明らかにし、AHI1遺伝子の重要性を示唆しています。また、疾患の分子メカニズムや臨床的特徴についても詳細な情報を提供しています。
遺伝子型と表現型の関係
さらに、Elsayedら(2015)は、c.3196C-T変異体をホモ接合性で保有する非症候群性難聴を分離する家系の非罹患者を報告しました。これにより、AHI1のC末端SH3ドメインが正常な発達には必要ないことが示されました。この結果から、疾患関連遺伝子の変異が必ずしも病原性を持つわけではなく、機能解析と分離解析を通じてその影響を評価する必要があることが強調されました。
また、Elsayedら(2015)は、妊娠前スクリーニングパネルに含まれるAHI1のバリアントを評価する重要性を指摘し、切断型バリアントであっても、その病原性を確認するために厳格な解析が必要であることを示唆しました。この研究は、遺伝子型と表現型の相関を明らかにし、遺伝的要因が疾患の発症にどのように影響するかを理解する上で貴重な情報を提供しています。

この記事の筆者:仲田洋美(医師)






