疾患概要
Ceroid lipofuscinosis, neuronal, 8 神経セロイドリポフスチン症8 600143 AR 3
神経細胞性セロイドリポフスチン症-8(CLN8病)は、私たちの遺伝情報が記された染色体の一つ、8番染色体の特定の位置(8p23)にあるCLN8という遺伝子に問題があることで発症します。この遺伝子に変異があると、体の正常な機能に必要なタンパク質がうまく作られないか、機能しなくなります。この病気は、遺伝子の変異が両親から受け継がれる場合(ホモ接合体変異)や、両親から異なる変異を受け継ぐ場合(複合ヘテロ接合体変異)に起こります。
また、「北欧てんかんバリアント」という特別な形のCLN8病もあります。これは、同じCLN8遺伝子の問題によるものですが、病気の症状や進行の仕方が異なるタイプの病気です。つまり、CLN8遺伝子の異なる変異が、体に異なる影響を与えることがあります。これらの情報は、病気を理解し、適切な治療法を見つけるために重要です。
神経細胞セロイドリポフスチン症(NCL、またはCLNとも呼ばれる)は、子どもや若者に影響を及ぼす致命的な神経変性疾患の群を指します。NCLは、その臨床的特徴と遺伝的背景の多様性から、非常に異質な疾患群として認識されています。この疾患群の共通点は、細胞内に特定の自家蛍光性リポ色素蓄積物質が蓄積することにより、超微細構造学的に異なるパターンを示すことです。
Mole et al. (2005)によれば、CLN8関連のNCLでは、細胞内に「顆粒状」、「曲線状」、「指紋状」のリポピグメントパターンが混在するのが特徴です。これらの特徴的なパターンは、NCLの診断において重要な手がかりとなります。
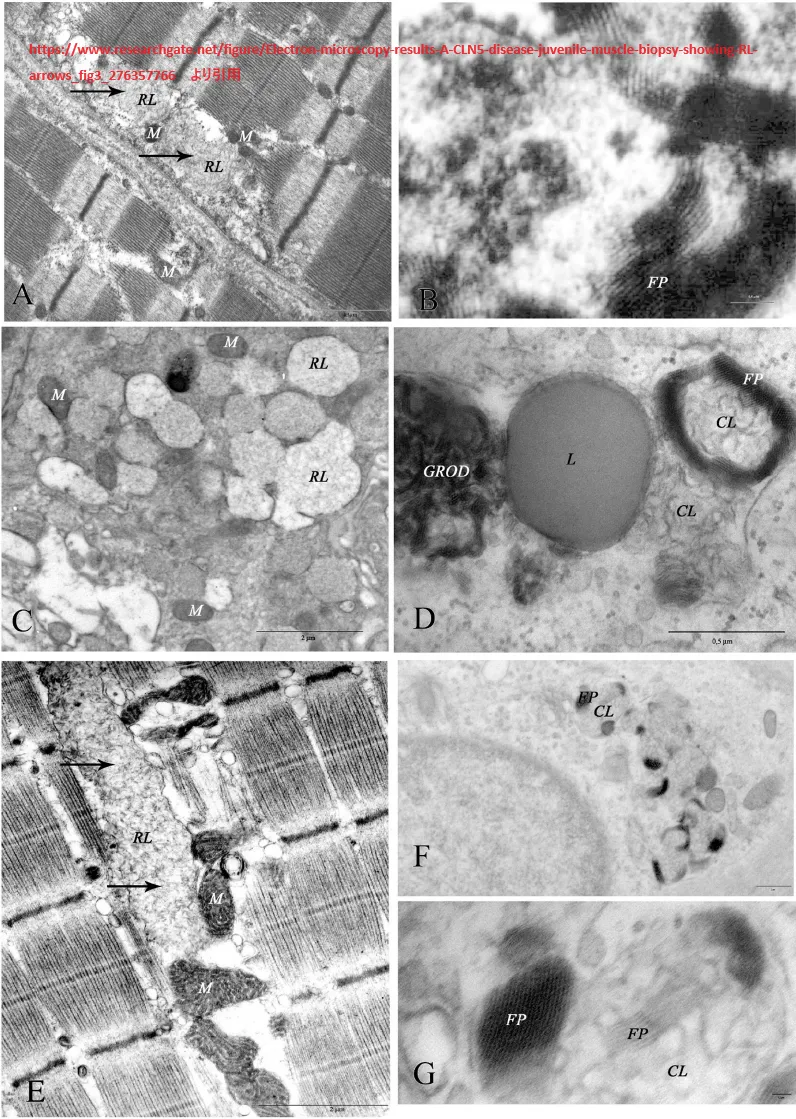
www.researchgate.net/figure/Electron-microscopy-results-A-CLN5-disease-juvenile-muscle-biopsy-showing-RL-arrows_fig3_276357766
より引用。電子顕微鏡の結果。A.CLN5病(幼若期):筋生検でRL(矢印)を示す(A. L. Taratuto氏提供)。B. 若年性CLN6病:筋生検でFPが密に詰まっている(提供:A. L. Taratuto)。C.若年性CLN7:エクリン分泌腺にRLを伴う小胞を示す皮膚生検。D. CLN8疾患(先天性):皮膚生検で、脂質滴(L)に隣接したGROD、CLおよびFPを示す。E. 若年性CLN3病:筋生検で、CLが主に筋小胞下コンパートメントに認められる(矢印);ミトコンドリアの肥大に注意(M)(提供:A. L. Taratuto)。F.若年性CLN3病:エクリン汗腺にFPとCLが混在する皮膚生検。G.若年性CLN3病(Fの詳細):FPとCLが混在した小体。略号: CLは曲線状小体、RLは直線状小体、FPは指紋状小体、GRODは顆粒状親水性沈着物、Mはミトコンドリオン、Lは脂質滴。
NCLは遺伝的に不均一であり、複数の遺伝子変異が関連していることが知られています。例えば、CLN1疾患は、CLN1遺伝子の変異によって引き起こされ、それぞれのNCLの形態は、特定の遺伝子変異と関連しています。これらの遺伝子変異は、神経細胞の機能障害と細胞死を引き起こし、結果として進行性の神経変性をもたらします。
NCLの臨床的表現型は、早期発症の視覚障害、認知機能の低下、運動機能障害、てんかん発作など、多岐にわたります。疾患の進行に伴い、患者はますます多くの神経系の機能障害を経験し、多くの場合、若年での死亡に至ります。
NCLの治療には、現在、症状を管理し、患者の生活の質を改善するための支援的な措置が中心となっています。しかし、この疾患群の遺伝的および生物学的な理解が深まるにつれて、より効果的な治療法の開発に向けた研究が進められています。
遺伝的不均一性
臨床的特徴
Wheelerら(1999): トルコ人の6家族が報告され、これらの家族は既知のCLN遺伝子座にマッピングされていませんでした。しかし、その後の研究で、4家族が染色体8p23上のCLN8遺伝子との連鎖が認められ、Rantaら(2004)によってこれらの家族の患者全員においてCLN8遺伝子の変異が同定されました。
Topcuら(2004): トルコ人患者28人中17人に後期幼児性CLNのトルコ変種が観察されました。これらの患者は、平均発症年齢が5.1歳で、てんかん発作や運動障害が最も一般的な症状でした。病気の進行とともに、精神退行、ミオクローヌス、言語障害、視力低下、人格障害などが発生しました。
Cannelliら(2006): 血縁関係のないイタリア人患者3例が報告され、これらの患者は精神運動発達の遅れ、てんかん発作、ミオクローヌス、進行性の視力低下を示しました。脳MRIでは大脳および小脳の萎縮が認められました。
Allenら(2012): 5.5歳のアイルランド人男児が報告され、早期のグローバルな発達遅滞、社会的引きこもり、進行するてんかん発作などが観察されました。脳MRIでは、白質に高輝度と小脳萎縮が認められました。
これらの研究は、CLN8遺伝子変異によって引き起こされる疾患の臨床的特徴についての理解を深めています。特に、てんかん発作、運動障害、精神運動発達の遅れ、進行性の視力低下などの特徴が、この疾患を特徴づける主要な臨床的現象として報告されています。また、これらの研究成果は、遺伝子診断の精度を高め、適切な治療法の開発に貢献する可能性があります。
マッピング
その後、Rantaらは2004年にこの研究をさらに進め、トルコの18家族を対象にした大規模な調査を行いました。この調査で、9家族ではCLN8遺伝子の変異と病気との関連が確認されましたが、残りの9家族では、この領域に共通の変異が見られなかったため、彼らの病気はCLN8遺伝子とは関係がないと考えられました。
このように、特定の遺伝子の変異が特定の病気の原因であることを突き止めることは、遺伝性疾患の研究において非常に重要です。これにより、病気の診断や治療法の開発に役立つ情報が得られます。
遺伝
この遺伝のパターンは、特定の疾患が家族内でどのように伝わるかを理解する上で非常に重要です。CLN8関連のNCLは、特定の遺伝子変異によって引き起こされるため、遺伝カウンセリングや家族計画において重要な情報を提供します。保因者である親から疾患が発症する子供へのリスクを評価することが可能になり、遺伝的検査を通じて家族内の他のメンバーのリスクも評価できます。
このように、Rantaらの研究はCLN8関連NCLの遺伝学的理解を深めることに貢献し、将来的な治療法の開発や予防策の策定に向けた基礎情報を提供しました。
分子遺伝学
●Rantaら(2004)
研究背景: Rantaらは、いわゆるトルコ型の遅発性小児CLNに関する研究を行いました。この研究では、CLN8遺伝子のホモ接合体変異を9家系の患者において同定しました。
変異の同定: 合計で4つのホモ接合体変異(607837.0002-607837.0004を参照)が同定されました。このうち4つは以前の研究(WheelerらとMitchellら)から、残りの5つはTopcuらの報告から同定されました。
疾患の特徴: これらの変異は、フィンランド北部てんかん変異型に対立する形でCLN8遺伝子と関連していることが明らかにされました。しかし、トルコ人患者においては、遺伝子型と表現型の間に明確な相関関係は見られませんでした。研究者らは、トルコ型の患者の表現型がフィンランド北部てんかん患者とは異なることを指摘しています。
●Cannelliら(2006)
研究背景: Cannelliらは、血縁関係のないイタリアのCLN8患者3例における研究を行いました。
変異の同定: これらの患者において、CLN8遺伝子のホモ接合または複合ヘテロ接合の変異が同定されました(607837.0005および607837.0006を参照)。
●結論
これらの研究成果は、CLN8遺伝子の変異が引き起こす遅発性小児CLNにおける遺伝子型と表現型の多様性を示しています。特に、地理的・民族的背景による遺伝子変異の分布の違いと、それが疾患表現型に与える影響についての理解を深めることができます。このような知見は、遺伝子診断や将来の治療法の開発において重要な役割を果たす可能性があります。
動物モデル
Bronson et al., 1993: この研究では、運動ニューロン変性(MND)マウスと呼ばれるNCLの自然発生モデルにおいて、Cln8遺伝子に1-bpの挿入(コドン90で267-268insC)が同定されました。この変異はフレームシフトを引き起こし、機能しないタンパク質の生成を予測しています。これはNCLの自然発生モデル動物における分子基盤の理解における重要な進歩を示しています。
Katz et al., 2005: イングリッシュ・セッター犬において、常染色体劣性遺伝のNCLがCln8遺伝子の特定の変異(leu164-to-pro、L164P)によって引き起こされることが同定されました。これは、犬を用いたNCLの遺伝的研究における重要な発見です。
Guo et al., 2014: オーストラリアン・シェパードとブルーヒーラーの混血犬において、Cln8遺伝子のヌクレオチドc.585でGからAへの転移が報告されました。この変異は、予想されるトリプトファンの代わりにナンセンス変異を引き起こし、生後8ヶ月からNCLに特徴的な神経学的徴候を示す原因となりました。この研究では、1,488のオーストラリアン・シェパードのDNAサンプルから、ヘテロ接合またはホモ接合の変異を持つ個体が発見され、その中の3頭はNCLの臨床症状を示し、2頭ではNCLが死後の脳組織評価により確認されました。
これらの研究は、NCLの分子基盤を理解し、将来的な治療法の開発に向けた基礎研究に貢献しています。動物モデルを使用することで、特定の遺伝子変異が疾患の発症にどのように関与しているかを解明することが可能になり、これは人間における同様の疾患の研究にも直接的な影響を与えます。







