疾患に関係する遺伝子/染色体領域
疾患概要
Carbamoylphosphate synthetase I deficiency カルバミルリン酸合成酵素Ⅰ欠損症 237300 AR 3
カルバモイルリン酸合成酵素I欠損症は、染色体2q34上に位置するCPS1遺伝子(608307)のホモ接合体または複合ヘテロ接合体変異によって引き起こされる常染色体劣性遺伝病です。この状態は尿素サイクルの最初のステップに影響を与え、体がアンモニアを尿素に変換する能力を低下させます。その結果、血中のアンモニア濃度が危険なレベルまで上昇し、高アンモニア血症を引き起こします。
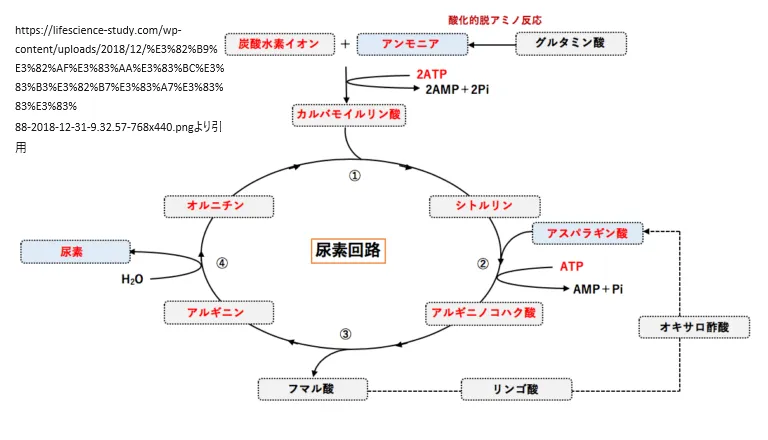
カルバモイルリン酸合成酵素I欠損症には、大きく分けて2つの型があります。
1. 致死的な新生児型:生後数日以内に症状が現れる最も重篤な形態です。高アンモニア血症による神経系の損傷が特徴で、未治療の場合、乳児期に死亡する可能性が高いです。この型では、異常な眠気、呼吸数や体温の調節不良、哺乳困難、嘔吐、異常な体動、てんかん発作、昏睡などの症状が見られます。
2. 重篤ではない遅発型:この形態は、生涯にわたり軽度の症状が出たり、ストレスや病気の際にのみ症状が顕著になることがあります。成人期まで診断されない場合もあり、症状は新生児型ほど重篤ではありませんが、適切な治療と管理が行われない場合、健康に深刻な影響を及ぼす可能性があります。
尿素サイクル障害は、脳症、呼吸性アルカローシス、および高アンモニア血症を特徴とし、カルバモイルリン酸合成酵素I欠損症の他にも、オルニチントランスカルバミラーゼ欠損症、アルギニノコハク酸合成酵素欠損症、シトルリン血症、アルギニノコハク酸リアーゼ欠損症、アルギナーゼ欠損症などがあります。
治療には、血中のアンモニア濃度を低減させる薬物療法、特別な食事療法、場合によっては血液浄化処置などが含まれます。早期診断と治療開始が、生存率や生活の質の向上に非常に重要です。患者と家族に対する遺伝学的カウンセリングも推奨されます。
カルバモイルリン酸合成酵素I欠損症は、尿素サイクル障害の一種であり、体内でアンモニアを安全な物質に変換するプロセスが妨げられる遺伝性の状態です。このプロセスは主に肝臓で行われ、アンモニアを尿素に変換して排出します。カルバモイルリン酸合成酵素I(CPS1)はこのプロセスの最初のステップを担い、その活動の不足はアンモニアの適切な処理を妨げます。この状態は、体内で生成されるアンモニアの過剰な蓄積を引き起こし、高アンモニア血症をもたらします。
アンモニアは神経毒性を持ち、特に脳に対して有害です。高アンモニア血症は、神経系に多くの問題を引き起こす可能性があり、特に新生児や乳児において重篤な症状や発達上の遅れを引き起こす可能性があります。生後数日以内に、カルバモイルリン酸合成酵素I欠損症の乳児は、高アンモニア血症の影響として、異常な眠気、呼吸や体温の調節不良、哺乳困難、嘔吐、異常な体動、てんかん発作、昏睡などの症状を示すことがあります。
治療には、アンモニアの血中濃度を低下させることが重要です。これは、薬物療法、特定のアミノ酸の制限を伴う特別な食事、時には血液浄化法(例えば、血液透析)を用いて行われます。早期診断と治療の開始は、潜在的に命を脅かす合併症を防ぎ、長期的な健康への影響を最小限に抑えるために重要です。
カルバモイルリン酸合成酵素I欠損症の患者は、生涯にわたり継続的な医療的監視と管理が必要となります。感染症やその他のストレスが高アンモニア血症の発作を引き起こす可能性があるため、これらの状態を迅速に治療し、適切な予防策を講じることが重要です。また、患者やその家族に対する教育と支援も、日常生活の管理と長期的なアウトカムの改善において重要な役割を果たします。
CPS1遺伝子に関連する変異は、カルバモイルリン酸合成酵素I(CPSI)欠損症の原因となります。これらの変異により、カルバモイルリン酸シンテターゼIの酵素が正常に機能しなくなります。酵素が短くなったり、形が不正になったり、または全く生成されなくなることがあります。
酵素の構造はその化学反応の調節能力に直接影響を与えます。したがって、カルバモイルリン酸シンテターゼI酵素の形状や機能が損なわれると、尿素サイクルでの重要な役割を果たすことができなくなります。尿素サイクルは、体内の余分な窒素を尿素に変換し排泄する過程であり、この過程が妨げられると、アンモニアとしての窒素が体内に蓄積します。アンモニアは神経系に対して特に有害であり、その結果、神経学的問題やカルバモイルリン酸合成酵素I欠損症に関連するその他の症状や徴候が引き起こされます。
臨床的特徴
早期発症型
先天性高アンモニア血症とカルバモイルリン酸合成酵素の活性低下を示す早期発症型のカルバモイルリン酸合成酵素I欠損症は、1970年にFreemanらによって初めて報告されました。この病態は、Hommesら(1969年)とEbels(1972年)によって報告された、重篤な大脳障害を持つ1人を含む3人の兄弟が罹患していた家族でも確認されました。Gelehrter and Snodgrass(1974年)は、この疾患で不足しているのがミトコンドリアのカルバモイルリン酸合成酵素であると述べました。
鈴木らは、1986年にCPS I欠損症の女性乳児について報告しました。この乳児は生後9日目に死亡し、CPS Iの酵素タンパク質もmRNA活性も検出できませんでした。
Finckhらは、1998年に重度のCPS I欠損症を持つ男性乳児を報告しました。この乳児は生後11日で死亡しました。彼は正期産の3日後に高アンモニア血性昏睡を発症しました。肝臓の病理組織学的検査では、びまん性微小小胞性脂肪症、明瞭な局所性肝細胞およびクッパー細胞のグリコーゲノーシス、および酵素的CPS I活性の不在が認められました。CPS1遺伝子の変異も検出されました(608307.0002)。
遅発型
遅発型カルバモイルリン酸合成酵素I欠損症は、成人期や幼少期の後期に発症する可能性がある希少な病態で、尿素サイクル障害の一種です。この疾患は、CPS1遺伝子の変異によりカルバモイルリン酸合成酵素Iの活性が不足し、体内でアンモニアを適切に処理できなくなることで特徴づけられます。遅発型は、重症度が異なり、症状の出現が遅れることがありますが、重篤な合併症を引き起こすリスクは依然として存在します。
Granotら(1986) の報告は、遅発型CPS欠乏症を示した9歳のアラブ人の小児のケースを紹介しています。この子供はReye症候群に似た症状を示し、高アンモニア血症による不可逆的な脳障害を負いました。
Verbiestら(1992) は、バルプロ酸を投与された後に高アンモニア血症を発症し、CPS I欠乏症と診断された32歳の女性について報告しています。この症例は、特定の薬剤が高アンモニア血症を引き起こすトリガーとなる可能性があることを示しています。
Wongら(1994) は、出産後に昏睡状態となりCPS I欠損症であることが判明した26歳の女性について述べています。このケースは、特定の生理的またはストレスの多い状況が尿素サイクル障害の潜在的な発症を引き起こすことがあることを強調しています。
Batshawら(2014) による尿素サイクル障害患者614人の分析では、カルバミルリン酸合成酵素欠損症が全体の約3%に見られ、死亡リスクが新生児期と遅発性の両方で42%であることが示されました。
これらのケーススタディは、遅発型カルバモイルリン酸合成酵素I欠損症がいかに予測不可能であり、しばしば通常の生活を送っている人々に突然発症するかを示しています。感染症や特定の薬剤(例えば、バルプロ酸)、さらには生理的ストレスの増加(例えば、出産)が発症のトリガーになり得ます。この疾患の管理と治療には、適切な診断、持続的な監視、および個別化された治療戦略が必要です。遅発型でも、高アンモニア血症の発作を避けるために、特定の食事制限やトリガーとなる条件からの保護が必要になることがあります。
その他の特徴
遺伝
McReynoldsらによる1981年の研究では、肝ミトコンドリアカルバモイルリン酸合成酵素欠損症が常染色体劣性遺伝することが明らかにされました。この病気に罹患している2人の姉妹の肝生検サンプルでは、酵素活性が正常値の約6%と大幅に低下していることが観察されました。一方で、病気に罹患していない正常な兄弟は酵素活性が正常値を示しています。また、症状が表れていない両親の酵素活性は中間の水準(正常値の32%と54%)でした。この結果は、両親がそれぞれ1つの変異遺伝子を持ち、この遺伝子を2つ受け継ぐことで病気が発症することを示唆しています。これにより、この疾患が常染色体劣性遺伝の特徴を持つことが証明されました。
頻度
原因
カルバモイルリン酸合成酵素I欠損症は尿素サイクル障害という遺伝性疾患のグループに属します。この状態では、カルバモイルリン酸合成酵素Iの活動が低下または欠如しており、尿素サイクルが正常に機能せず、窒素が毒性の少ない尿素として排泄される代わりに、有毒なアンモニアの形で血液中に蓄積します。アンモニアは脳にとって特に有害であり、過剰なアンモニアは神経系に悪影響を及ぼし、カルバモイルリン酸合成酵素I欠損症に伴う神経学的問題やその他の症状を引き起こす可能性があります。
治療・臨床管理
Brusilowらは1984年に、カルバモイルリン酸合成酵素欠損症、オルニチントランスカルバミラーゼ欠損症、及びシトルリン血症を持つ小児におけるエピソード性高アンモニア血症の治療成功を報告しました。治療には、安息香酸ナトリウム、フェニル酢酸ナトリウム、アルギニンの静脈内投与、窒素を含まない静脈栄養、および他の手段が効果を示さない場合の透析が含まれていました。
分子遺伝学
分子遺伝学におけるCPS I欠損症について、Hoshideらは1993年に新生児の日本人女児からCPS1遺伝子にホモ接合性のミスセンス変異を発見しました。さらに、Kurokawaらは2007年にCPS I欠損症と診断された18人の日本人患者のうち16人からCPS1遺伝子に25個の異なる変異、その中には19個の新規変異が含まれていることを確認しました。発症は13歳と31歳という遅い年齢である2人の患者も含まれており、遺伝子型と表現型の間には明確な相関が見られませんでした。
Haberleらは2011年に、24年間にわたって収集された205人のCPS I欠損症患者の組織とDNAサンプルを分析し、192個の異なる病原性変異を同定しました。これらには130個の新規変異が含まれており、これまでに報告された変異を合わせると、ほとんどの変異は各家系に固有のものであることが明らかになりました。再発変異の多くはCpGジヌクレオチドで起こる傾向があり、特にミスセンス変異の大部分は、酵素の触媒部分をコードする領域やその他の重要なドメインに集中していました。大腸菌の酵素を用いた比較モデリングは、これらミスセンス変異の位置が進化的に重要であり、タンパク質のフォールディングに影響を与える可能性があることを示唆しています。
CPS I欠損症は、体内でアンモニアを尿素に変換する過程に関わる酵素であるカルバモイルリン酸合成酵素 I (CPS I)の活動不足によって生じます。この病状はアンモニアの血中濃度の異常上昇を引き起こし、重篤な健康問題を引き起こす可能性があります。
遺伝子型と表現型の関係
両患者の遺伝子解析では、2つのスプライス部位変異(608307.0010および608307.0011)が見つかり、これらは複合ヘテロ接合体であることが明らかにされました。さらに、大腸菌を用いたクローニング実験から、これらの患者の対立遺伝子発現の割合に違いがあることが示されました。より重篤な症状を示した孫は、一方の変異が他方の3倍の割合で発現していました。この発現の偏りが臨床症状の違いをもたらしている可能性がありますが、その具体的なメカニズムは未解明です。Klausらは、この発現の偏りが家系内での臨床的ばらつきの原因の一つであると結論付けています。
集団遺伝学
CPS I欠損症(カルバモイルリン酸合成酵素I欠損症)は、尿素サイクル障害の一種であり、尿素サイクルが正常に機能しないことにより、アンモニアなどの有害な物質が体内に蓄積する病気です。これらの推定は、この希少な遺伝性疾患の発生頻度を理解するための重要な情報を提供します。
疾患の別名
Carbamyl-phosphate synthetase I deficiency disease
Congenital hyperammonemia, type I
カルバモイルリン酸合成酵素I欠損症
カルバモイルリン酸合成酵素I欠損症
I型先天性高アンモニア血症







