疾患に関係する遺伝子と表現型
常染色体劣性栄養障害型表皮水疱症:COL7A1
栄養障害型反対型表皮水疱症:COL7A1
限局型栄養障害型表皮水疱症:COL7A1
表皮水疱症、常染色体劣性遺伝、修飾因子:MMP1
疾患概要
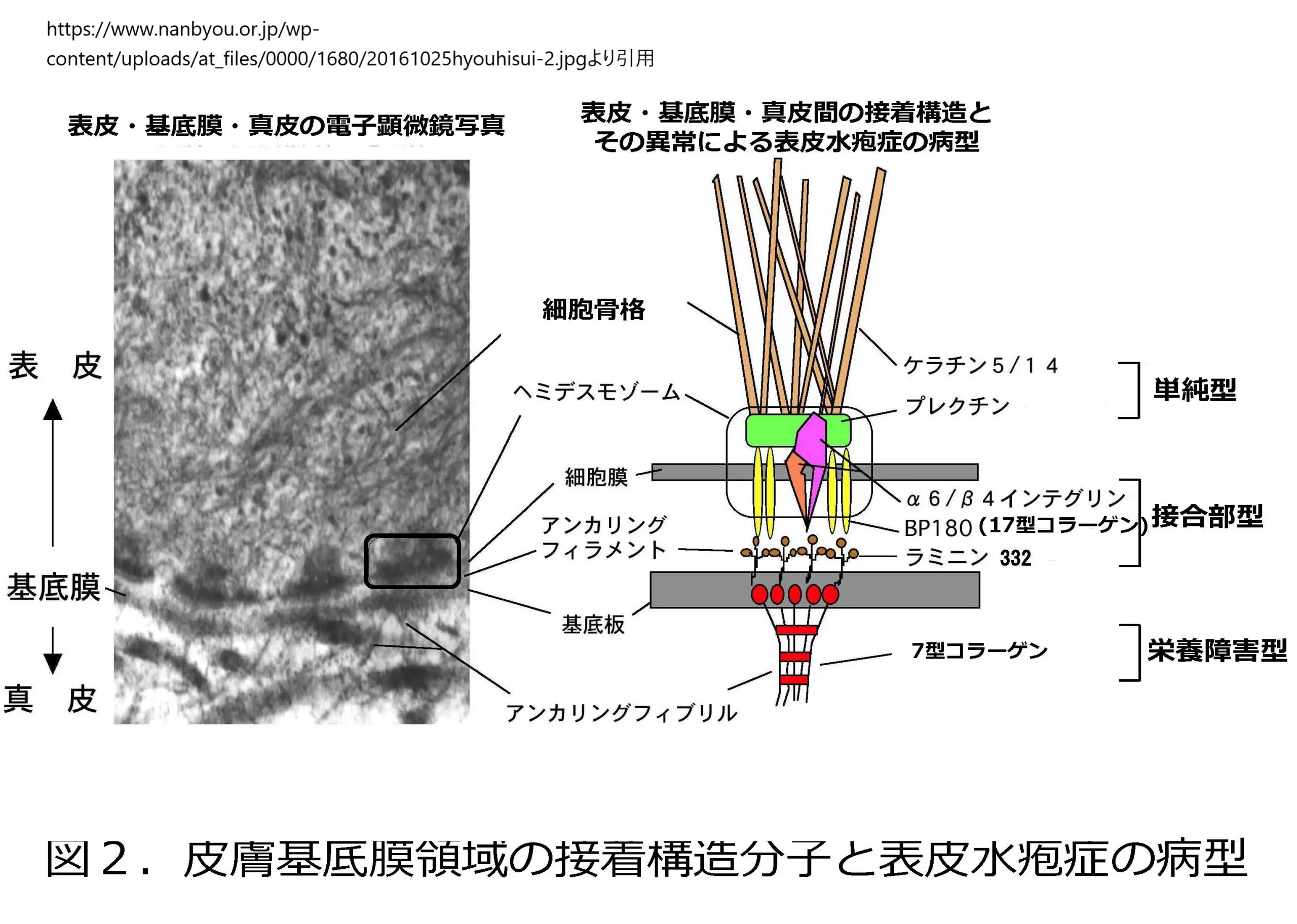
常染色体劣性遺伝性表皮水疱症(Recessive Dystrophic Epidermolysis Bullosa, RDEB)および限局性表皮水疱症は、COL7A1遺伝子の変異によって引き起こされる皮膚疾患です。この遺伝子は染色体3p21に位置し、VII型コラーゲンの生成に必要な指示を提供しています。VII型コラーゲンは皮膚の基底膜ゾーンにおけるアンカー線維の形成に重要な役割を果たしており、その変異は皮膚の強度と弾力性の低下を引き起こします。
●RDEBの特徴
RDEBは出生時から発症する重篤な状態で、基底膜下レベルでの皮膚の水疱形成が特徴です。これらの水疱は繰り返し発生し、手足や関節の瘢痕化や拘縮を引き起こします。また、粘膜病変による消化管の狭窄も発症し得るため、栄養不良に陥ることがあります。さらに、RDEB患者は侵攻性扁平上皮癌の発症リスクが高くなります。
●アレリックな疾患
RDEBと関連するアレリック疾患には、常染色体優性DEB (DDEB)がありますが、DDEBの表現型はRDEBほど重篤ではありません。また、非症候性先天性爪疾患-8 (NDNC8)は、RDEB家系の中でヘテロ接合体保因者に常染色体優性形質として現れることがあります。
RDEBは、その重篤な臨床的特徴と生涯にわたる管理が必要な疾患であるため、早期診断と対症療法が重要です。現在、遺伝子治療や皮膚移植など、この疾患の治療法を改善するための研究が進行中です。これらの研究は、RDEB患者の生活の質を向上させる可能性があります。
https://www.nanbyou.or.jp/wp-content/uploads/at_files/0000/1680/20161025hyouhisui-2.jpg
より引用
栄養障害型表皮水疱症(Dystrophic Epidermolysis Bullosa, DEB)は、7型コラーゲン(COL7A1遺伝子によってコードされる)の異常により引き起こされる遺伝性の皮膚疾患です。7型コラーゲンは、皮膚の基底膜と真皮を接着する重要な役割を持つアンカー線維の主要成分であり、このタンパク質の構造や機能の異常は、皮膚の構造的安定性に重大な影響を及ぼします。
病態メカニズム
7型コラーゲンの異常があると、基底膜と真皮の間の正常な接着が損なわれ、微小な外傷や摩擦などの軽度の物理的刺激によっても、基底膜と真皮の間に水疱が形成されます。この水疱形成は、皮膚の脆弱性を著しく増加させ、繰り返しの水疱とその治癒過程による瘢痕形成、爪の異常や関節の拘縮など、患者の生活の質に大きく影響する一連の臨床症状を引き起こします。
臨床的特徴
DEBの患者は出生時、またはそれに近い時期から皮膚の脆弱性を示し、最小限の外傷で水疱が形成されます。水疱は自然にまたは刺激によって破れ、治癒する際に瘢痕を残すことが一般的です。長期にわたる疾患の進行は、皮膚の広範な瘢痕化、変形、機能障害を引き起こし、場合によっては食道狭窄などの深刻な合併症につながることがあります。
診断と治療
DEBの診断は、臨床的特徴、家族歴、皮膚生検による組織学的検査、および遺伝子検査に基づいて行われます。現在、DEBに対する根治的な治療法は存在しませんが、対症療法、感染予防、栄養管理、外科的介入による合併症の管理などが行われます。また、遺伝子治療や細胞治療など、新たな治療法の開発が進められています。
DEBは、その重篤な影響と管理の複雑さから、患者やその家族にとって大きな負担となる疾患です。継続的な研究と治療法の改善により、将来的には患者の生活の質の向上と疾患の進行の抑制が期待されます。
臨床的特徴
●3人の日本人兄弟の症例
出生時からの皮膚の極端な脆弱性
広範な瘢痕形成、爪の欠損、指の癒合、関節拘縮
口腔粘膜の水疱形成や食道狭窄
重篤な栄養不良と貧血
長兄は21歳で死亡
皮膚生検では基底膜下の真皮-表皮剥離が確認され、アンカー線維が存在しないことが示された
血縁関係のない4家族の症例
出生時または出生後すぐに指、口唇、口腔粘膜、耳などで水疱形成
高齢になると、多発性びらん、瘢痕形成、癒合による手のミトン変形、関節拘縮が認められる
爪の欠損や食道の狭窄
電子顕微鏡検査では、萎縮性EBと一致する低形成アンカリング線維と硬膜下レベルでの裂開が確認された
Bart症候群と表現型的に重複する症例があった
偏性ヘテロ接合体の両親は臨床的に影響を受けていない
これらの報告は、DEBの臨床的スペクトラムの広さとその重篤な影響を示しています。DEBの診断、管理、および治療は、患者およびその家族にとって重要な課題であり、患者の生活の質の向上と疾患の進行の抑制に焦点を当てた継続的な医療が必要です。このような症例報告は、DEBの理解を深め、将来的な治療法の開発に貢献する可能性があります。
常染色体劣性遺伝性表皮水疱症のinversa亜型
常染色体劣性遺伝性表皮水疱症のinversa亜型は、手足の指を除き、主に体の屈曲部に病変が現れる珍しい疾患の一種です。Gedde-Dahlは1971年に、ノルウェー人6家系13人におけるEBD inversaの最初の報告を行い、Hallopeau-Siemens型DEBと比べて、皮膚病変の分布や角膜、肛門周囲、眼窩周囲の病変の違いを指摘しました。橋本らも1976年に、2人の姉妹におけるこの疾患の報告をしています。
PearsonとPallerは1988年に、重度の口腔および食道粘膜病変の発生を特徴とする4人のアメリカ人DEB inversa患者を報告しました。足の爪は軽度から中等度の萎縮を示していましたが、顕微鏡的変化は表皮水疱症のHallopeau-Siemens型に類似していると述べられています。
Wrightらは1993年に、RDEB inversaの10名の患者を報告しました。この診断は硬膜下の組織剥離と典型的な屈曲部に限局した水疱形成によって確認されました。皮膚病変の重症度や分布にはばらつきがありましたが、Hallopeau-Siemens型DEBに特徴的な顕著な趾節腫、重度の全身性水疱形成、成長遅延を示す患者はいませんでした。全例に口唇病変がみられ、口唇閉鎖症や舌乳頭の欠損などが観察されました。
Hovnanianらは1994年に、血縁関係のない2例の劣性DEB inversaを報告しました。11歳の女児は、頚部、腋窩、鼠径部、口腔に水疱形成が見られ、重度の再発性食道狭窄を有していました。皮膚生検では、硬膜下の裂開や正常な固定線維の欠如などが確認されました。
Linらは1995年に、栄養障害型表皮水疱症の2例を報告しました。1症例では、外科的矯正を必要とする指網瘢痕と軽度の合指症が見られました。
これらの報告から、inversa型表皮水疱症は、その特有の病変分布と臨床症状により、他の表皮水疱症の亜型と区別されます。この疾患の理解と診断には、これらの特徴的な症状を詳細に調査することが重要です。
その他の特徴
Travisらの1992年の分析によると、表皮水疱症患者246人のうち、劣性ジストロフィー型EB患者の76%、優性ジストロフィー型EB患者の20%、接合型EB患者の15%、単純型患者の2%が嚥下障害を経験していました。栄養障害型表皮水疱症では特に舌癒着や小ストーマがみられ、劣性型では優性型の8倍の頻度で発生していました。食事の外傷や減少によって誘発されたこれらの病変は、便秘と栄養失調を悪化させました。食道の狭窄は頻繁に発生し、単数または複数の食道ウェブを伴っていました。
Bassらによる1993年の報告では、早産女性の母親が妊娠中に血清および羊水中のα-フェトプロテイン(AFP)濃度が顕著に上昇し、羊水アセチルコリンエステラーゼバンドが陽性であることが報告されましたが、超音波検査は陰性でした。
Bourkeらは1995年に、劣性DEBの2人の姉妹が致死的な全身性アミロイドーシスを発症したことを報告しました。1人は22歳で診断され、皮膚感染が広範囲に及んでいたため透析が不可能でした。姉は26歳でアミロイドーシスの検索で陰性だったが、35歳でアミロイド腎症を発症しました。
Melvilleらによる1996年の報告では、2人の無関係な小児が致死的な拡張型心筋症を発症しました。これらの小児は重度の栄養不良と発育遅延を示し、心筋症の原因は微量栄養素の欠乏、特にセレン欠乏である可能性が高いと示唆されました。
マッピング
Hovnanianらによる1992年の研究では、染色体3p21に位置するCOL7A1遺伝子が劣性栄養障害型表皮水疱症と連鎖していることが証明されました。この発見は、19の有益な家系を用いた分析により、最大lodスコア3.95で確認されました。lodスコアは、特定の遺伝子と疾患の間の連鎖が偶然よりも高い確率で存在することを示す統計的尺度です。この研究により、COL7A1遺伝子の位置が明らかになり、劣性栄養障害型表皮水疱症の遺伝的基盤に関する理解が深まりました。
また、Ryynanenら(1991)とUittoら(1992)による研究では、COL7A1遺伝子のPvuII RFLP(制限断片長多型)が優性DEB(表皮水疱症)と連鎖していることが証明されました。これらの研究により、優性DEBと劣性DEBが同一の遺伝子、すなわちCOL7A1遺伝子の異なる突然変異によって引き起こされる可能性が示唆されました。この発見は、遺伝子の変異が疾患の形態にどのように影響するかを理解する上で重要な一歩です。
これらの研究成果は、遺伝子マッピングが遺伝性疾患の研究においていかに重要であるかを示しています。遺伝子の正確な位置を特定することで、遺伝子変異がどのようにして特定の疾患の原因となるのか、またその疾患がどのように遺伝するのかを理解するための基盤が築かれます。このような知識は、遺伝性疾患の診断、予防、治療に役立つ可能性があります。
診断
Anton-Lamprechtらによる1981年の研究では、胎児鏡を使用して胎児の皮膚を直接観察し、さらに皮膚生検のサンプルを電子顕微鏡で検査することで、Hallopeau-Siemens型の表皮水疱症ジストロフィー(重度の劣性ジストロフィック表皮水疱症)の出生前診断に成功しました。これは、疾患の早期発見と対応計画の策定において非常に重要なステップです。
また、Hovnanianらによる1995年の研究では、COL7A1遺伝子の解析を用いて全身性劣性DEBの出生前診断が実施されました。この技術は、6家族における再発リスクがある胎児に対して行われ、5家族では重度のHallopeau-Siemens型、1家族では汎発性の非変異型の症状が確認されました。羊水細胞から得られた胎児のDNA解析を通じて、これらの胎児が少なくとも1つの正常なCOL7A1対立遺伝子を受け継いでいることが明らかにされました。
これらの進歩は、遺伝子解析技術の進展とともに、遺伝性疾患の出生前診断がより正確に、かつ早期に行えるようになったことを示しています。親や医療提供者が適切な対応を準備する上で、これらの診断方法は非常に貴重です。
治療・臨床管理
Chenら(2002年)は、自己不活性化最小レンチウイルスベクターを用いてヒト皮膚細胞にCOL7A1遺伝子を導入し、VII型コラーゲンの持続的な合成と分泌を達成しました。この遺伝子修正細胞を用いて免疫不全マウス上でヒト皮膚を再生させることに成功し、VII型コラーゲンの発現とアンカー線維の形成が生体内で回復されることを示しました。
Ortiz-Urdaら(2002年)は、インテグラーゼベースの遺伝子導入技術を使用して、RDEB患者の細胞にCOL7A1のcDNAを安定的に組み込みました。この技術により、再生された皮膚はVII型コラーゲンの発現やアンカリング線維の形成など、RDEBの症状を安定的に改善しました。
Wagnerら(2010年)は、栄養障害型表皮水疱症(EB)の小児に対して、免疫除去化学療法後の同種造血幹細胞移植を実施しました。この治療により、移植後に水疱形成の減少が認められ、一部の患者では臨床的に顕著な改善が見られましたが、移植片拒絶反応や感染症のリスクも報告されています。
Guideら(2022年)は、栄養障害型表皮水疱症(DEB)患者を対象に、COL7A1を送達する単純ヘルペスウイルス1型ベースの遺伝子治療薬B-VECの有効性を検証しました。この研究では、B-VECを局所投与した創傷の大部分で完全治癒が観察され、プラセボ投与群と比較して有意に高い治癒率を示しました。
これらの研究は、遺伝子治療が特定の遺伝性皮膚疾患の治療において有望なアプローチであることを示しています。レンチウイルスベクターやインテグラーゼベースの遺伝子導入技術、同種造血幹細胞移植、ウイルスベースの遺伝子治療薬など、様々な手法が検討されており、それぞれの手法が持つリスクと利益を考慮した上で、患者に最適な治療法を選択することが重要です。
分子遺伝学
Christianoら(1995)は、常染色体劣性遺伝のDEBを持つ3人の日本人兄弟で、COL7A1遺伝子の2つの切断型変異(120120.0005; 120120.0006)の複合ヘテロ接合体を発見しました。罹患していない両親はそれぞれの変異をヘテロ接合体で持っていました。
Christianoら(1996)は、RDEB(再発性表皮水疱症)を発症した4家族の罹患者で、COL7A1遺伝子のグリシン置換変異を同定しました。2つの家系では、グリシン置換と早期終結変異の複合ヘテロ接合体が見られましたが、他の2つの家系ではグリシン置換のホモ接合体が見られました。これらの変異は、疾患症状を示さないヘテロ接合体保因者ではサイレントでした。
清水ら(1999)は、八田らによって以前に報告されたRDEB患者で、COL7A1遺伝子のG2316R(120120.0042)とG2287R(120120.0023)の2つの変異の複合ヘテロ接合体を特定しました。G2287Rの変異を持つヘテロ接合体保因者は、非症候性の孤立性爪ジストロフィーを有していましたが、皮膚は正常でした。
Sato-Matsumuraら(2002)は、RDEBを持つ日本人2家系を研究し、足の爪に限局したジストロフィーを示すが、皮膚の脆弱性は認められない家系で、グリシン置換G1595R(120120.0024)とG1815R(120120.0025)のヘテロ接合体をそれぞれ特定しました。これらの変異は、限定的な爪の変形を引き起こし、COL7A1のナンセンス変異やフレームシフト変異と組み合わさることで、皮膚の脆弱性に寄与する可能性があります。
Varkiら(2007)は、栄養障害型表皮水疱症患者310人のCOL7A1遺伝子を解析し、重度の劣性DEB患者は切断型変異を、より軽度の優性DEB患者はグリシン置換を持つ傾向があることを指摘しました。一部の患者は優性変異と劣性変異の両方を持っていました。
Hovnanianら(1994)は、劣性DEB inversaと古典的RDEBを持つ2人の患者で、COL7A1遺伝子のヘテロ接合体変異(R109X; 120120.0040)を特定しました。この疾患が劣性遺伝性であることを示す説得力のある証拠を提示しました。
Kahoferら(2003)は、劣性DEB inversaの2人の兄弟で、COL7A1遺伝子の2つの変異(120120.0041; 120120.0045)の複合ヘテロ接合体を特定しました。これらの研究は、COL7A1遺伝子の変異が表皮水疱症の様々な形態にどのように関与しているかを示しており、特にグリシン置換変異がヘテロ接合体では無症状であるか、または優性遺伝性のDEBを引き起こす可能性があることを明らかにしています。
修飾遺伝子
MMP1(コラゲナーゼ)遺伝子の変異や活性の変化は、栄養障害型表皮水疱症(DEB)の発症および病態に重要な役割を果たしています。VII型コラーゲンはコラゲナーゼによって容易に分解されるため、この酵素の活性の変化はDEBの臨床症状に大きく影響を及ぼすことが示されています。
Bauer(1977)による研究では、DEB患者から得られたプロコラゲナーゼが、通常のものと比較して熱分解性が高く、カルシウム親和性が低く、in vitroでの活性が低いことが見出されました。このことから、コラゲナーゼの構造遺伝子の変異、酵素の翻訳後修飾の欠陥、またはコラゲナーゼの正常な分解を制御する遺伝子の変異が考えられました。
一方、BauerとEisen(1978)の研究では、DEB患者の大部分において、培養された皮膚線維芽細胞が過剰にコラゲナーゼを産生していることが観察されました。これは、DEBの病理にコラゲナーゼの亢進が関与していることを示唆しています。しかし、Winbergら(1989)の研究では、RDEB患者の一部にのみコラゲナーゼの過剰発現が認められ、この酵素の発現亢進がすべてのDEB患者に一様に見られるわけではないことが示されました。
Titeuxら(2008)の研究では、MMP1遺伝子の多型(rs1799750)がDEBの重症度と有意な関連を持つことが見出されました。この多型がコラゲナーゼ活性を増加させることにより、より重篤な表現型につながることが示され、MMP1がDEBの修飾遺伝子である可能性が示唆されました。
これらの研究成果は、DEBの発症および進行におけるコラゲナーゼの重要性を浮き彫りにし、DEBの病態理解や治療法開発において重要な示唆を与えています。特に、MMP1遺伝子の多型がDEBの臨床症状に与える影響についての知見は、個々の患者に対するより適切な治療戦略の選択や、新たな治療法の開発に貢献する可能性があります。
遺伝子型と表現型の関係
この研究で注目すべきは、既知の19のミスセンス変異全てが、アルギニンまたはグリシンの置換であったことです。アルギニン置換の5個の変異のうち3個(例:R2063G)、そしてグリシン置換の14個の変異のうち9個(例:G1907E)は、RDEBの逆位型に特異的であることが示されました。
この発見は、遺伝子型と表現型の相関を理解する上で非常に重要です。RDEB inversaは、COL7A1遺伝子の特定のミスセンス変異に強く関連しており、これらの変異がどのようにして疾患の特異的な表現型を引き起こすかについての理解を深めることができます。また、特定のアミノ酸の置換が疾患の特定の形態にどのように影響を与えるかについての洞察を提供し、将来の研究や治療戦略の開発に役立つ情報を提供します。
動物モデル
Fritschらによる2008年の研究では、Col7a1遺伝子の発現を条件付きで不活性化するトランスジェニックマウスモデルが開発されました。このマウスモデルは、正常なCol7a1レベルの約10%しか発現しない低形成個体を生み出しました。ホモ接合体のマウスは生まれた時は正常でしたが、出生後24〜48時間以内に前足に水疱が形成されました。栄養不良のため全身状態が悪化し、舌にも水疱ができましたが、流動食を与えることで生存率が向上しました。前足のミトン型変形は、創傷治癒過程における異常な線維化と軟部組織の収縮によるものであることが明らかにされました。このマウスモデルの表現型は、皮膚の脆弱性、爪の異常(爪ジストロフィー)、偽指症、成長遅延など、ヒトの劣性遺伝病に類似していました。野生型線維芽細胞を皮内に注入することで、Col7a1の沈着と機能が回復し、マウスの表現型が改善されることが示されました。
これらの研究は、遺伝性表皮水疱症の理解を深め、将来的な治療法の開発に向けた基礎的な知見を提供しています。
歴史
疾患の別名
DYSTROPHIC EPIDERMOLYSIS BULLOSA, AUTOSOMAL RECESSIVE
EPIDERMOLYSIS BULLOSA DYSTROPHICA, HALLOPEAU-SIEMENS TYPE; EBR1
EPIDERMOLYSIS BULLOSA DYSTROPHICA, GENERALIZED SEVERE, AUTOSOMAL RECESSIVE
●Other entities represented in this entry:
EPIDERMOLYSIS BULLOSA DYSTROPHICA, AUTOSOMAL RECESSIVE, LOCALISATA VARIANT, INCLUDED
EPIDERMOLYSIS BULLOSA DYSTROPHICA INVERSA, AUTOSOMAL RECESSIVE, INCLUDED
別名;記号
常染色体劣性遺伝性栄養障害型表皮水疱症
表皮水疱症; EBR1
常染色体劣性全身性重症表皮水疱症
この項目に含まれる他の疾患
表皮水疱症、常染色体劣性、限局性バリアント、含む
常染色体劣性遺伝、栄養障害型反対型表皮水疱症に含まれるもの







