目次
神経系に関わる様々な疾患の中で、CACNA1A遺伝子は特に重要な役割を果たしています。この遺伝子は脳内のカルシウムチャネルをコードしており、神経伝達に必須の機能を持ちます。CACNA1A遺伝子の変異は、片頭痛、てんかん、運動失調症など多様な神経症状を引き起こすことが知られています。
この記事では、CACNA1A遺伝子の基本情報から関連疾患、遺伝子検査の意義まで詳しく解説します。神経発達に関わる症状や家族歴のある方々にとって、理解を深める一助となれば幸いです。
CACNA1A遺伝子の基本情報
CACNA1A遺伝子(Calcium Channel, Voltage-Dependent, P/Q Type, Alpha-1A Subunit)は、19番染色体(19p13.13)に位置し、P/Q型電位依存性カルシウムチャネル(CaV2.1)のα1Aサブユニットをコードしています。このチャネルは主に脳内、特に小脳のプルキンエ細胞に多く発現しており、神経伝達物質の放出に重要な役割を果たしています。
プルキンエ細胞とは:
プルキンエ細胞は、小脳皮質に存在する大型の神経細胞で、複雑に分岐した樹状突起と特徴的な構造を持ちます。小脳回路における唯一の出力細胞であり、運動の協調性や姿勢制御、バランス維持、運動学習など、小脳の重要な機能を担っています。これらの細胞は特にCACNA1A遺伝子が豊富に発現しており、その機能障害は運動失調や不随意運動などの症状と関連しています。脊髄小脳失調症6型(SCA6)などのCACNA1A遺伝子関連疾患では、このプルキンエ細胞の変性や脱落が観察されます。
CACNA1A遺伝子の特徴:
- 遺伝子座位:19p13.13
- 遺伝子サイズ:約300kb
- エクソン数:47個
- コードするタンパク質:P/Q型カルシウムチャネルのα1Aサブユニット
- 発現部位:主に脳(小脳、大脳皮質、視床、視床下部)
電位依存性カルシウムチャネルは、興奮性細胞へのカルシウムイオンの流入を媒介するだけでなく、筋収縮、ホルモンや神経伝達物質の放出、遺伝子発現などのカルシウム依存性プロセスにも関与しています。CACNA1A遺伝子から生成されるタンパク質は、神経細胞間の情報伝達において中心的な役割を担っているのです。
CACNA1A遺伝子の構造と機能ドメイン
CACNA1A遺伝子は、約300キロベースの長さを持ち、47個のエクソンから構成される大型の遺伝子です。この遺伝子からコードされるP/Q型カルシウムチャネルタンパク質は、複雑な構造を持つ膜貫通タンパク質で、その構造は機能と密接に関連しています。
CACNA1A遺伝子がコードするCaV2.1チャネルの基本構造:
- 4つの反復ドメイン(I-IV):各ドメインは類似した構造を持ち、膜を貫通する部分を形成
- 各ドメインの6つの膜貫通セグメント(S1-S6):特にS4セグメントは電位センサーとして機能
- P-ループ領域:S5とS6の間に存在し、イオン選択性フィルターを形成
- 細胞内N末端とC末端:調節タンパク質との相互作用や翻訳後修飾の部位
- ドメイン間リンカー:ドメイン間を結ぶ細胞内ループで、チャネルの調節に関与
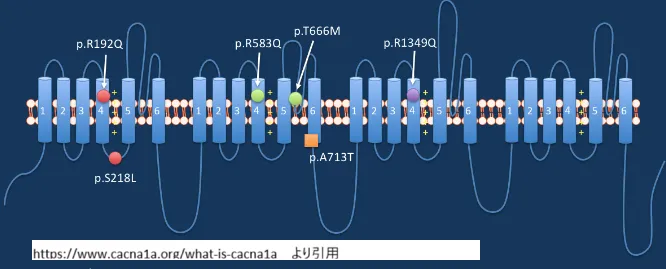
この複雑な構造により、CACNA1A遺伝子がコードするCaV2.1チャネルは、神経細胞の活動電位に応じて開閉し、カルシウムイオンの細胞内流入を精密に制御することができます。特にC末端領域には、カルモジュリン結合部位やポリグルタミン領域などの重要な機能部位が含まれています。
CACNA1A遺伝子のスプライシングバリアント
CACNA1A遺伝子は選択的スプライシングにより複数のアイソフォームを生成します。特にエクソン37には2つの選択的バリアント(37aと37b)が存在し、これらは神経伝達物質放出に異なる影響を与えることが知られています。また、エクソン47内のCAGリピート長の違いもタンパク質の特性に影響を与えます。
CACNA1A遺伝子変異と構造-機能連関:
CACNA1A遺伝子変異の位置は、症状の表現型に大きく影響します。例えば:
- 電位センサー領域(S4セグメント)の変異:FHM1に関連し、チャネルの活性化電位を変化させる
- ポア領域(P-ループ)の変異:イオン選択性や透過性に影響し、FHM1やEA2を引き起こす
- C末端のCAGリピート伸長:ポリグルタミン鎖の蓄積によりSCA6を引き起こす
- フレームシフト/ナンセンス変異:機能喪失型変異としてEA2に関連する
CACNA1A遺伝子の構造解析は、関連疾患の病態メカニズム理解や標的治療法開発において重要な役割を果たしています。特に、特定のドメインを標的とした薬剤開発や遺伝子治療は、将来の治療戦略として期待されています。
CACNA1A遺伝子の機能と働き
CACNA1A遺伝子がコードするP/Q型カルシウムチャネルは、神経シナプスにおいて特に重要な働きをしています。このチャネルは、活動電位が神経終末に到達すると開口し、カルシウムイオンの流入を促進します。この流入が神経伝達物質の放出を引き起こし、神経間の情報伝達が行われるのです。
また、CACNA1A遺伝子からは、完全長のカルシウムチャネルタンパク質だけでなく、CACNA1A C末端ポリペプチド(α-1ACT)も生成されます。このタンパク質は転写因子として機能し、小脳の発達に関わっています。これは内部リボソーム進入部位(IRES)の使用によって生成され、特に小脳の正常な発達において重要な役割を果たしていることが研究で明らかになっています。
カルシウムチャネルの分類と主な機能:
- L型:筋収縮、ホルモン分泌、遺伝子発現
- N型:神経伝達物質の放出
- P/Q型(CACNA1A):シナプス伝達、小脳機能
- T型:神経細胞の興奮性調節
- R型:様々な神経機能
CACNA1A遺伝子の機能異常は、カルシウムシグナリングの乱れを引き起こし、神経細胞の興奮性に変化をもたらします。これが様々な神経疾患の基盤となるメカニズムと考えられています。
CACNA1A遺伝子関連疾患
CACNA1A遺伝子の変異は、いくつかの特徴的な神経疾患と関連しています。これらの疾患は「カルシウムチャネル病」とも呼ばれ、同一の遺伝子変異が様々な臨床症状を引き起こす「表現型の多様性」が特徴です。
1. 家族性片麻痺性片頭痛1型(FHM1)
家族性片麻痺性片頭痛1型(FHM1、OMIM: 141500)は、CACNA1A遺伝子のミスセンス変異によって引き起こされる常染色体優性遺伝疾患です。主な特徴として、片側の麻痺を伴う前兆(視覚障害、感覚障害など)と激しい頭痛発作が挙げられます。
FHM1の特徴的な症状:
- 片側の一時的な麻痺(通常30分から数時間持続)
- 視覚前兆(閃輝暗点など)
- 激しい頭痛(数時間から数日間持続)
- 嘔吐、光過敏、音過敏などを伴うことがある
- 軽度の頭部外傷後に症状が誘発されることがある
多くの場合、FHM1は小児期から青年期に発症し、家族内での浸透率は高いものの、発作の頻度や重症度には個人差があります。一部の患者さんでは、発作時に意識障害や昏睡を伴う重症例も報告されています。
【重要】S218L変異と頭部外傷リスクについて
FHM1に関連する変異の中で、特に注意が必要なのが「S218L変異」です。この変異を持つ患者さんは、日常の些細な頭部外傷が引き金となり、明らかな意識清明期を経た後に致死的な大脳浮腫や持続的な昏睡、早期のてんかん発作を引き起こす重大なリスクが報告されています。
動物モデルを用いた研究では、異常な神経興奮の波(皮質拡延性抑制:CSD)が皮質だけでなく、より深い脳の構造にまで反響的に伝播してしまうことが確認されています。この変異が確認された場合、コンタクトスポーツを避けるなど、日常生活における厳重な頭部保護が極めて重要となります。
▼ 疾患の詳細と関連する検査についてはこちら
2. 発作性失調症2型(EA2)
発作性失調症2型(EA2、OMIM: 108500)は、CACNA1A遺伝子の機能喪失型変異(ナンセンス変異、フレームシフト変異、スプライシング変異など)によって引き起こされる常染色体優性遺伝疾患です。主な特徴は、数時間から数日間持続する一過性の小脳性運動失調発作です。
EA2の特徴的な症状:
- 突然発症する一過性の失調(歩行障害、協調運動障害)
- 眼振(眼球の不随意運動)
- 構音障害(ろれつが回らない)
- めまい、吐き気、嘔吐
- 発作間欠期にも軽度の小脳症状が残存することがある
- アセタゾラミドによる治療が有効なことがある
EA2の発作は、ストレス、運動、アルコール摂取、カフェイン摂取などによって誘発されることがあります。患者さんの約半数では、加齢に伴い小脳萎縮が進行し、発作間欠期にも持続的な小脳症状が見られるようになります。
3. 脊髄小脳失調症6型(SCA6)
脊髄小脳失調症6型(SCA6、OMIM: 183086)は、CACNA1A遺伝子のエクソン47に存在するCAGリピートの異常伸長(通常21-30リピート)によって引き起こされる常染色体優性遺伝性疾患です。この疾患は、緩徐進行性の小脳性運動失調を主徴とします。
SCA6の特徴的な症状:
- 緩徐進行性の歩行失調
- 四肢の協調運動障害
- 構音障害
- 眼振
- 通常40-50歳代で発症
- MRIでの小脳萎縮
SCA6は比較的遅い発症年齢と緩やかな進行が特徴で、生命予後への影響は他のSCAタイプと比較して小さいとされています。しかし、症状の進行により日常生活動作に大きな支障をきたすことがあります。
パラダイムシフト:SCA6は「チャネル機能異常」ではなく「転写因子病」
近年の研究により、SCA6の病態メカニズムに大きなパラダイムシフトが起きました。CACNA1A遺伝子は、細胞膜のカルシウムチャネルを作るだけでなく、遺伝子の途中(IRES:配列内リボソーム進入部位)から、全く別の「α1ACT」という核内転写因子を独立して作り出す「二重翻訳機構」を持っています。
SCA6では、変異によって異常に長く伸びたポリグルタミン鎖を持つ「変異型α1ACT」が作られ、これが細胞の核内に蓄積して強い毒性を発揮します。つまり、SCA6はチャネルの働きが悪くなる病気ではなく、この毒性タンパク質が小脳の神経細胞(プルキンエ細胞)を直接的に死滅させることによって起こるのです。
【専門コラム】なぜSCA6は「わずかなCAGリピート伸長」で発症するのか?
ハンチントン病をはじめとする多くの「ポリグルタミン病(CAGリピート病)」では、病的になるためには通常36〜40回以上の長いリピート伸長が必要です。しかし、SCA6ではわずか「19〜33回」という非常に短い伸長で小脳失調を発症します。この特異な現象の背景には、SCA6独自のメカニズムがあります。
- タンパク質全体のサイズとの比率: 異常の舞台となる「α1ACT」は、分子量約75kDaと比較的小さなタンパク質断片です。巨大なタンパク質(ハンチンチンなど)の一部が伸びる疾患とは異なり、小さなα1ACTにおいては、数個のグルタミン(CAG)の追加であっても、タンパク質全体の立体構造を決定的に破壊してしまいます。
- ダイレクトな核内毒性: α1ACTは、小脳プルキンエ細胞の核へ移動し、直接DNAに結合して神経発生を制御する「転写因子」です。非常にデリケートな機能を持つため、わずかな構造変化でも本来の標的(GRN遺伝子など)に結合できなくなるだけでなく、核内ですぐに有毒な塊(凝集体)を形成し、直接的に細胞死を引き起こしてしまいます。
このように、変異が起きる「場所(転写因子としてのペプチド)」の特殊性が、SCA6における発症閾値の低さを説明する重要な鍵となっています。
【専門コラム】なぜ変異は「プルキンエ細胞」だけを選択的に破壊するのか?
CACNA1A遺伝子の変異は全身の細胞に存在しますが、SCA6では小脳の「プルキンエ細胞」だけが選択的にダメージを受けます(選択的脆弱性)。この理由は、α1ACTの特異な働きにあります。
- 小脳での圧倒的な生成量: α1ACTを作り出す「IRES(配列内リボソーム進入部位)」の活性は、小脳で極めて高いことが分かっています 。そのため、毒性を持つ変異型α1ACTが小脳に大量に蓄積してしまいます 。
- 生存に関わるネットワークの要: α1ACTは転写因子として、神経細胞の生存や成長に不可欠な遺伝子群(GRN、PMCA2など)を直接コントロールしています 。巨大で複雑なプルキンエ細胞はこれらの遺伝子群への依存度が他よりも圧倒的に高いため、α1ACTの異常が直ちに細胞の死結びついてしまうのです 。
CAGリピート数の判定基準:SCA6とハンチントン病の比較
SCA6におけるCAGリピートの異常伸長は、他の代表的なポリグルタミン病であるハンチントン病と比較して、非常に短いリピート数で病原性(完全浸透)を示すことが臨床上の大きな特徴です。発症前診断や遺伝カウンセリングにおいては、以下の基準値に基づいた慎重な評価が行われます。
| 判定区分 | 脊髄小脳失調症6型 (SCA6) |
ハンチントン病 (HD) |
|---|---|---|
| 正常アレル | 18回 以下 | 26回 以下 |
| 中間アレル (次世代で伸長するリスクあり) |
– (※SCA6では極めて稀) |
27 〜 35回 |
| 不完全浸透アレル (発症しない場合もある) |
19 〜 20回 | 36 〜 39回 |
| 完全浸透アレル (原則として生涯のどこかで発症する) |
21 〜 33回 | 40回 以上 |
このように、SCA6では「21回以上」という、ハンチントン病であれば完全に「正常」と判定されるような短いリピート数であっても、完全浸透(生涯のどこかで発症する可能性が極めて高い状態)となります。
ただし、完全浸透アレルであっても、リピート数が比較的短い場合(例えば21〜23回など)は、発症年齢が遅くなる(50〜60代以降)傾向があります。そのため、発症前診断でこの帯域の結果が出た場合、ご本人の長期的なライフプランニングと、専門医による継続的な心理的サポート・遺伝カウンセリングが非常に重要となります。
▼ SCA6の詳しい疾患解説と確定診断(検査)についてはこちら
4. 発達性てんかん性脳症42型(DEE42)
発達性てんかん性脳症42型(DEE42、OMIM: 617106)は、CACNA1A遺伝子の特定の変異によって引き起こされる重篤な神経発達障害です。新生児期や乳児期早期からのてんかん発作と発達遅滞が特徴です。
DEE42の特徴的な症状:
- 生後早期からの難治性てんかん発作
- 重度の発達遅滞
- 筋緊張低下または亢進
- 脳波異常
- 進行性の神経学的症状
DEE42は稀な疾患であり、現在も研究が進行中です。適切な診断と早期介入が重要とされています。
▼ 関連する遺伝子パネル検査についてはこちら
遺伝子型と表現型の関係
CACNA1A遺伝子関連疾患では、変異の種類と臨床症状(表現型)との間に一定の関連性が認められています。
機能獲得(GOF)と機能喪失(LOF)のダイナミクス:
CACNA1A遺伝子変異は、カルシウムチャネルの働きを「過剰」にするか「低下」させるかによって、全く異なる症状を引き起こします。
- 機能獲得型(GOF)変異(主にFHM1): チャネルが通常より低い電位で開いたり、閉じるのが遅くなったりします。これにより過剰なカルシウムが流入し、神経伝達物質(グルタミン酸など)が過剰放出され、脳のネットワークが異常な興奮状態に陥ります。
- 機能喪失型(LOF)変異(主にEA2): ナンセンス変異などによりチャネルが機能しなくなったり、カルシウムを通しにくくなったりします。小脳プルキンエ細胞でカルシウム流入が不足すると、細胞のペースメーカー機能が損なわれ、運動失調や眼振が現れます。
- 混合型変異とてんかん(DEE): 一部の変異は、GOFとLOFの両方の特徴を併せ持つ「混合型」のプロファイルを示します。小児の重篤なてんかん性脳症では、単なるチャネルの変化というよりも、それが引き起こす脳全体の「興奮と抑制のバランス(E/Iインバランス)」の破綻が重症度を決定づけると考えられています。
しかし、同じ変異であっても、個人間や家族間で症状の重症度や表現型に違いがあることも多く、他の遺伝的要因や環境要因の影響も考慮する必要があります。また、一部の患者さんでは、複数の症状(例:FHM1とSCA6の併発)がみられることもあります。
これらの複雑な遺伝子型-表現型相関は、CACNA1A遺伝子が神経系の発達と機能において多面的な役割を担っていることを示唆しています。
CACNA1A遺伝子検査の重要性
CACNA1A遺伝子検査は、関連する神経疾患の診断を確定するために重要な役割を果たします。特に、家族歴のある片頭痛、原因不明の運動失調症、発作性神経症状、乳幼児期発症のてんかんなどの症状がある場合に考慮されます。
遺伝子検査が有用な状況:
- 家族性片麻痺性片頭痛が疑われる場合
- 発作性または進行性の小脳症状がある場合
- 原因不明のてんかん(特に早発性)がある場合
- 家族内に複数の神経疾患患者がいる場合
- 治療法の選択や予後予測に役立てたい場合
当クリニックでは、CACNA1A遺伝子を含む様々な神経疾患関連遺伝子の検査を提供しています。特に、片頭痛NGSパネル、晩発性失調症NGSパネル、GABA代謝障害NGSパネル、神経伝達物質代謝障害NGSパネル、てんかん包括的NGSパネルなどにCACNA1A遺伝子が含まれています。
CACNA1A遺伝子検査が含まれる主な検査パネル:
- 片頭痛NGSパネル
- 晩発性失調症NGSパネル
- GABA代謝障害NGSパネル
- 神経伝達物質代謝障害NGSパネル
- てんかん包括的NGSパネル
- シングル遺伝子検査(CACNA1A単独検査)
遺伝子検査の結果は、疾患のメカニズム理解、治療方針の決定、家族計画、予防措置の実施などに役立ちます。また、家族の他のメンバーが同様の症状を発症するリスクの評価にも有用です。
遺伝学的検査の推奨事項と最新技術
神経発達障害や神経症状の診断において、遺伝学的検査は重要な役割を果たします。CACNA1A遺伝子関連疾患の診断も、適切な遺伝学的検査によって確定することができます。
各専門機関の遺伝学的検査に関する推奨事項:
- 米国医学遺伝学会(ACMG)は、発達遅延、知的障害、先天異常に対して、ゲノムまたはエクソーム解析を第一選択の検査とすることを推奨しています。
- 米国遺伝カウンセラー協会(NSGC)も、原因不明のてんかんを有するすべての患者に対して、ゲノムまたはエクソーム解析を第一選択検査とすることを推奨しており、このガイドラインは米国てんかん学会(AES)にも支持されています。
通常のエクソーム解析やゲノム解析で診断がつかなかった場合でも、最新の検査技術によって原因を特定できる可能性があります。特にCACNA1A遺伝子のようなスプライシングバリアントを持つ遺伝子の解析には、RNA解析が有効な場合があります。
最新技術:RNA統合シークエンス解析(RNA-ISE)検査
RNA統合シークエンス解析(RNA-ISE)検査は、通常のwhole exomeやwhole genome解析で診断がつかなかった方の約20%程度が、疾患原因を特定できるようになる革新的な技術です。この検査は、DNAレベルでは検出が難しいスプライシング異常や発現量の変化を検出できるため、CACNA1A遺伝子関連疾患を含む様々な遺伝性疾患の診断に役立ちます。
神経発達障害や神経症状が疑われる場合、まずは遺伝カウンセリングを受けて、適切な検査方法について相談することをお勧めします。CACNA1A遺伝子関連疾患が疑われる症状がある場合は、当クリニックの専門医にご相談ください。
CACNA1A遺伝子関連疾患の治療アプローチ
CACNA1A遺伝子関連疾患の治療は、現在のところ対症療法が中心となります。疾患特異的な治療法と一般的な支援策について解説します。
薬物療法
家族性片麻痺性片頭痛(FHM1):
- 発作予防:カルシウムチャネル遮断薬(ベラパミルなど)、トピラメート、ラモトリギンなど
- 急性期治療:NSAIDs、トリプタン系薬剤(一部患者に有効)
- 重症発作時:抗てんかん薬、ステロイド薬
発作性失調症(EA2):
- アセタゾラミド(発作頻度や重症度を減少させる効果がある)
- 4-アミノピリジン(カリウムチャネル遮断薬、発作予防に有効との報告がある)
- 抗てんかん薬(ラモトリギン、レベチラセタムなど)
脊髄小脳失調症6型(SCA6):
- 現時点で病気の進行を止める特異的治療はない
- アセタゾラミド(一部患者での症状改善報告あり)
- リハビリテーション(理学療法、作業療法、言語療法など)
発達性てんかん性脳症(DEE42):
- 抗てんかん薬(個別に有効性を評価)
- ケトン食療法(一部患者で有効との報告あり)
- 早期介入療法(理学療法、作業療法、言語療法など)
支援と管理
これらの疾患は慢性的な経過をたどることが多いため、適切な支援と管理が重要です:
- 定期的な神経学的評価と経過観察
- 症状に応じたリハビリテーション
- 日常生活の適応支援(補助具の使用など)
- 心理的サポート
- 発作誘発因子の回避(ストレス管理、適切な睡眠習慣など)
- 家族への教育と支援
CACNA1A関連疾患の管理において重要なポイント:
- 個々の症状に合わせた治療計画の立案
- 多職種による包括的アプローチ(神経内科医、小児神経科医、リハビリ専門家など)
- 定期的な評価と治療計画の見直し
- 発作誘発因子の特定と回避
- 生活の質(QOL)の維持・向上を目指した支援
将来的には、CACNA1A遺伝子の機能や変異の影響をより深く理解することで、より効果的な治療法や予防法の開発が期待されています。
CACNA1A遺伝子研究の最新動向
CACNA1A遺伝子に関する研究は現在も活発に行われており、病態メカニズムの解明や新たな治療法の開発が進められています。
動物モデルを用いた研究
マウスモデルを用いた研究では、CACNA1A遺伝子変異が神経細胞の機能や小脳の発達に与える影響が詳細に調べられています。特に、皮質拡延性抑制(CSD)という現象(片頭痛の前兆の原因と考えられている)との関連や、神経伝達物質放出の異常などが明らかになってきています。
遺伝子治療・薬物開発
カルシウムチャネルの機能を特異的に調節する薬剤の開発や、遺伝子治療の可能性についても研究が進んでいます。特に、SCA6に対するポリグルタミン凝集抑制剤や、FHM1に対する新規カルシウムチャネル調節薬などが注目されています。
精密医療への応用
患者さん個々のCACNA1A遺伝子変異タイプに基づいた治療選択(精密医療)も研究されています。例えば、特定の変異タイプに特化した薬剤の効果予測や、個別化された予防戦略の開発などが進められています。
今後の研究課題:
- 変異タイプと表現型の多様性を説明するメカニズムの解明
- カルシウムチャネル機能調節による神経保護効果の検証
- 遺伝子治療や RNA 治療法の開発
- バイオマーカーの同定と治療反応性の予測
- 他の遺伝子との相互作用の解明
これらの研究の進展により、将来的にはCACNA1A遺伝子関連疾患に対するより効果的な治療法や予防法が開発されることが期待されています。
次世代プレシジョン・メディシン(個別化医療)の最前線
長らく対症療法が中心だったCACNA1A関連疾患ですが、現在は「病気の根本原因に直接介入する」画期的なプレシジョン・メディシンの開発が急速に進んでおり、歴史的な転換期を迎えています。
てんかん(DEE)に対する画期的な新薬「Bexicaserin(LP352)」
難治性の発達性およびてんかん性脳症(DEE)に対し、現在最も期待されているのが「Bexicaserin(ベキシカセリン)」です。この薬は中枢神経のセロトニン受容体に選択的に働きかけ、脳の過剰な興奮を抑える新しいメカニズムを持っています。
最近完了した臨床試験(PACIFIC試験)では、プラセボ群と比較して約60%という劇的な発作頻度の減少を示し、多くの患者さんで発作が半減しました。現在、承認に向けた大規模な第3相試験が進行中です。
根本治療を目指す遺伝子治療と核酸医薬
変異した遺伝子そのものや、そこから作られる毒性物質を直接標的とする最先端の治療法も、臨床応用に向けて動き出しています。
- スプリットAAVベクター技術: 機能喪失型変異に対しては正常な遺伝子を導入する治療が理想ですが、CACNA1A遺伝子は大きすぎて従来のウイルスベクター(AAV)に入りきりません。そこで、長大な遺伝子を分割して細胞内に運び、中で再構築させる技術の開発が進んでいます。
- アンチセンス・オリゴヌクレオチド(ASO): 機能獲得型変異やSCA6の毒性タンパク質に対しては、ASOと呼ばれる核酸医薬を用いて、異常なタンパク質の設計図(mRNA)を分解し、生成を根本から防ぐ研究が進行中です。
- IRES選択的阻害薬: SCA6の治療において、生命維持に必須な正常チャネルには影響を与えず、毒性のあるα1ACTを作り出す「IRES」の働きだけを選択的にブロックする化合物の同定にも成功しつつあります。
CACNA1A遺伝子と関連疾患に関するよくあるご質問
- 家族がSCA6と診断されました。私も今は無症状ですが、将来発症するかどうかを調べる遺伝子検査(発症前診断)は受けられますか?
- はい、技術的には発症前診断は可能です。SCA6は常染色体優性遺伝のため、親が患者さんの場合、お子さんには50%の確率で遺伝します。
ただし、SCA6は現時点で発症を完全に予防したり進行を止めたりする根本治療が確立されていません。そのため、「もし将来発症することが確定した場合、その結果をどう受け止め、どうライフプランを立てるか」について、検査前に臨床遺伝専門医や遺伝カウンセラーと十分に時間をかけて話し合う(遺伝カウンセリング)ことが極めて重要です。当クリニックでも、ご本人の意思を最優先に慎重にサポートを行っています。
- 同じCACNA1A遺伝子の異常なのに、「片頭痛」になる人と「重いてんかん」になる人がいるのはなぜですか?
- 遺伝子の「どこに、どのような変異が起きたか」によって、カルシウムチャネルの働き方が全く異なるためです。
例えば、チャネルが過剰に働いてしまう変異(GOF)では片頭痛(FHM1)が起きやすく、逆にチャネルがうまく働かなくなる変異(LOF)では運動失調(EA2)が起きやすくなります。さらに、重篤なてんかん性脳症(DEE)を引き起こす場合は、この両方の特徴が混ざり合っていたり、脳全体の「興奮と抑制のバランス」が大きく崩れたりしていることが原因と考えられています。変異ごとの緻密なメカニズム解析が、現在の研究の最前線です。
- 子供がCACNA1A関連の発達性てんかん性脳症(DEE)と診断されました。記事にある新薬(Bexicaserin)はすぐに使えるようになりますか?
- Bexicaserin(ベキシカセリン)は、現在実施中の第1b/2a相試験で劇的な発作減少効果を示しており、非常に有望な新薬候補です。しかし、現在はまだ安全と効果を最終確認する大規模な「第3相国際共同治験」の段階であり、日本を含め、明日からすぐに一般の病院で処方できるわけではありません。
この第3相試験は2026年後半の完了を見込んでおり、その結果をもって各国の規制当局(FDAやPMDAなど)に承認申請が行われます。実用化まではもう少し時間がかかりますが、治療の未来は確実に明るくなっています。
- 家族性片麻痺性片頭痛(FHM1)と診断されています。「頭部外傷に気をつけて」と言われましたが、どの程度気をつければよいですか?
- FHM1の中でも、特に「S218L」という特定の遺伝子変異をお持ちの場合は、極めて厳重な注意が必要です。この変異がある場合、サッカーのヘディングや、軽く頭をぶつけた程度の「些細な頭部外傷」であっても、その後に致死的な脳のむくみ(大脳浮腫)や昏睡を引き起こすリスクがあることが報告されています。
主治医にご自身の変異のタイプを正確に確認し、もしリスクが高い変異であれば、コンタクトスポーツを避ける、自転車では必ずヘルメットを着用するなど、日常生活での徹底した頭部保護をお願いします。
まとめ
CACNA1A遺伝子は、様々な神経疾患(家族性片麻痺性片頭痛、発作性失調症、脊髄小脳失調症6型、発達性てんかん性脳症など)と関連しており、神経系の正常な機能において重要な役割を果たしています。
遺伝子検査は、これらの疾患の正確な診断、適切な治療選択、家族計画などにおいて重要な役割を果たします。また、研究の進展により、将来的にはより効果的な治療法や予防法の開発が期待されています。
片頭痛、運動失調、てんかんなどの症状がある方、特に家族歴がある場合は、CACNA1A遺伝子を含む遺伝子検査について医療機関にご相談ください。適切な診断と管理が、症状の軽減と生活の質の向上に役立つ可能性があります。
ミネルバクリニックでの遺伝子検査について:
ミネルバクリニックでは、臨床遺伝専門医が常駐し、様々な神経疾患に対する遺伝子検査を提供しています。CACNA1A遺伝子を含む検査パネルについてのご質問や相談は、お気軽にお問い合わせください。検査前後のサポートも充実しており、検査結果の解釈や今後の対応についても丁寧に説明いたします。
参考文献・出典
本記事は、CACNA1A遺伝子および関連疾患に関する最新の医学的知見および臨床試験データに基づき作成されています。さらに詳しい情報については、以下の主要な文献および臨床試験データをご参照ください。
- S218L変異と頭部外傷リスクについて:
- SCA6のパラダイムシフト(二重翻訳機構・α1ACT)について:
- 機能獲得(GOF)と機能喪失(LOF)・てんかん性脳症について:
- 次世代プレシジョン・メディシン(Bexicaserin・遺伝子治療)について:
- Bexicaserin for the treatment of seizures in developmental and epileptic encephalopathies (PACIFIC Phase 1b/2a Clinical Trial)
- ClinicalTrials.gov: A Study to Investigate LP352 in Children and Adults With Developmental and Epileptic Encephalopathies (DEE)
- National Ataxia Foundation: Identifying FDA-approved molecules to treat SCA6







