目次
8q24欠失症候群(ランガー・ギーディオン症候群)とは?
症状・原因・診断・治療を臨床遺伝専門医が解説
Q. 8q24欠失症候群(ランガー・ギーディオン症候群)とはどのような病気ですか?
A. 8番染色体長腕(8q24.11-q24.13)の微小欠失により、骨格・外胚葉・神経系に多彩な症状が現れる極めて稀な隣接遺伝子症候群です。
毛髪・鼻・指節骨症候群II型(TRPS II)とも呼ばれ、TRPS1・EXT1・RAD21の3遺伝子のハプロ不全により、特徴的顔貌・多発性外骨腫・知的発達遅延の3徴が生じます。
-
➤
原因 → 8番染色体8q24.11-q24.13領域の微小欠失(約3〜10Mb) -
➤
3徴(トライアド) → 特徴的顔貌・疎毛(TRPS1由来)+多発性外骨腫(EXT1由来)+知的発達遅延(RAD21関与) -
➤
遺伝形式 → 常染色体顕性(優性)遺伝、ただし大多数は新生突然変異 -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
頻度 → 100万人に1人未満の極めて稀な疾患(世界で約100例報告)
1. 8q24欠失症候群(ランガー・ギーディオン症候群)とは|基本情報
【結論】 8q24.11-q24.13欠失症候群(ランガー・ギーディオン症候群/TRPS II)は、8番染色体長腕の8q24.11から8q24.13領域が欠失する極めて稀な隣接遺伝子症候群です。世界で約100例しか報告されておらず、100万人に1人未満の頻度と推定されています。
「お子さんの顔つきや骨格が気になる」「検査で8q24欠失が見つかった」という方は、この病気について正確な情報を知ることが大切です。この症候群は複数の遺伝子が同時に欠失する「隣接遺伝子症候群」であり、骨格・外胚葉・神経系に多彩な症状が現れます。
💡 用語解説:「隣接遺伝子症候群」とは?
隣接遺伝子症候群(Contiguous Gene Deletion Syndrome)とは、染色体上で隣り合って存在する複数の遺伝子が同時に欠失することで、それぞれの遺伝子の機能喪失による症状が複合的に現れる疾患です。ランガー・ギーディオン症候群では、TRPS1・EXT1・RAD21という機能が異なる3つの遺伝子が一括して欠失するため、多彩な症状を呈します。
8q24欠失症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 8q24.11-q24.13欠失症候群(OMIM #150230) |
| 別名 | ランガー・ギーディオン症候群(LGS)、毛髪・鼻・指節骨症候群II型(TRPS II) |
| 原因 | 8q24.11-q24.13領域の微小欠失(約3〜10Mb) |
| 頻度 | 100万人に1人未満(世界で約100例報告) |
| 遺伝形式 | 常染色体顕性(優性)遺伝(大多数は新生突然変異) |
| 責任遺伝子 | TRPS1、EXT1、RAD21(主要3遺伝子) |
| 生命予後 | 基本的に正常(健常人と同等) |
歴史的背景と命名の由来
本症候群の概念は、2人の放射線科医の功績に由来します。1969年、Andres GiedionとLeonard Langerがそれぞれ独立して、毛髪・鼻・指節骨症候群の特徴に加えて多発性の外骨腫を有する症例を報告しました。これらの報告が統合される形で「ランガー・ギーディオン症候群」という名称が定着しました。
⚠️ TRPS I型との違い
TRPS I型はTRPS1遺伝子単独の点変異または遺伝子内微細欠失が原因で、特徴的顔貌や毛髪・爪の異常、円錐状骨端が見られますが、多発性外骨腫は伴いません。一方、TRPS II型(本症候群)はEXT1遺伝子を含むより広範な欠失であるため、多発性外骨腫が出現するのが最大の鑑別点です。
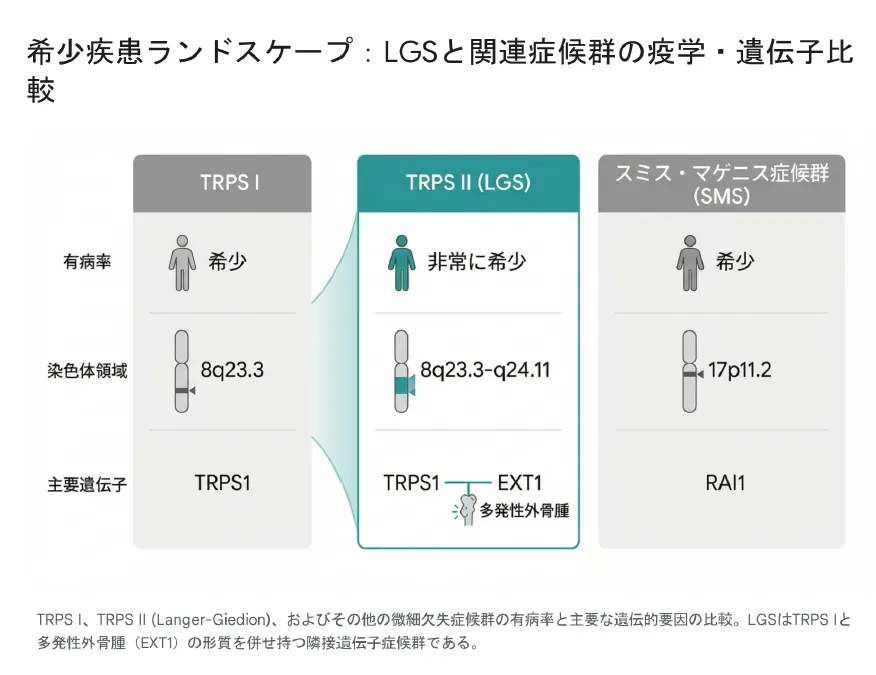
▲ TRPS I型、TRPS II型(LGS)、スミス・マゲニス症候群の疫学・遺伝子比較
2. 8q24欠失症候群の主な症状
【結論】 本症候群の症状は特徴的顔貌・多発性外骨腫・知的発達遅延の3徴(トライアド)を中心に、骨格系・外胚葉系・神経系に多岐にわたります。欠失の大きさにより症状の重症度が異なり、RAD21を含む広範な欠失ではより重度の知的障害を伴う傾向があります。
診断の3徴(トライアド)
① 特徴的顔貌・疎毛(TRPS1由来)
- •
梨状の鼻(鼻梁・鼻尖が球根状)
- •
薄い上唇、長い人中
- •
大きく突出した耳介
- •
疎毛(細く色素が薄い毛髪)
② 多発性外骨腫(EXT1由来)
- •
長管骨の骨幹端に多発性の骨軟骨腫
- •
生後1ヶ月〜6歳頃に出現
- •
思春期に急速に増大
- •
骨端線閉鎖で成長停止
③ 知的発達遅延(RAD21関与)
- •
軽度〜中等度の知的障害が多い
- •
正常知能の症例も存在
- •
実用的生活スキルはIQより良好
- •
RAD21欠失で重症化傾向
頭蓋顔面および外胚葉所見
-
•
鼻の形態:「梨状の鼻」(鼻梁と鼻尖が幅広く肉厚、球根状)、鼻翼は低形成
-
•
人中と口唇:人中は長く平坦、上唇の赤唇部は非常に薄い
-
•
耳介:大きく頭部から突出(立ち耳)、付着位置が低い場合も
-
•
眉毛:内側が濃く太く、外側に向かって急激に薄くなる
-
•
その他:小頭症(半数以上)、小顎症、高口蓋
-
•
頭髪:疎毛(細く、色素が薄く、成長が遅い)
-
•
進行性脱毛:特に男性で思春期以降に急速な脱毛(若年で完全脱毛も)
-
•
爪の異常:薄く脆い、スプーン状爪、異栄養症
-
•
皮膚:乳児期に皮膚弛緩(成長とともに改善傾向)
骨格系の異常
骨格系の異常は、本症候群で最も医学的管理を要する領域であり、患者さんの日常生活動作(ADL)やQOLに直接的な影響を与えます。
| 骨格所見 | 詳細 | 臨床的意義 |
|---|---|---|
| 多発性外骨腫 | 長管骨・肩甲骨・肋骨・骨盤・指骨に多発 | 疼痛、可動域制限、神経圧迫 |
| 円錐状骨端 | 手指中節骨・基節骨の骨端が円錐状に変形 | 短指症、巧緻運動障害 |
| 短指症 | 指骨の長軸方向への成長阻害 | 弯指、手指機能低下 |
| 低身長 | 出生前から成長遅滞(SGA) | GH欠乏の可能性を評価 |
| 股関節形成不全 | ペルテス病様変化、高頻度で合併 | 若年で人工関節置換の可能性 |
| 関節の経時的変化 | 小児期は硬直→成人期は弛緩 | 易脱臼性、外傷への脆弱性 |
⚠️ 外骨腫の悪性化リスク:成人期において外骨腫が軟骨肉腫へと悪性転化するリスクがあります。遺伝性多発性外骨腫(HME)における生涯リスクは約2〜5%とされ、LGSでも同様のリスクが推測されますが、文献上では「極めて稀」と報告されています。外骨腫の急激な増大や疼痛の変化があれば精査が必要です。
泌尿生殖器系の合併症
近年の症例集積により、本症候群は単なる骨・外胚葉疾患にとどまらず、内臓系、特に泌尿生殖器系に重大な形成異常を伴う全身性疾患であることが認識されています。
-
•
膣閉鎖/処女膜閉鎖:膣が形成されていない、または膜で閉じている状態
-
•
子宮留血腫:月経血が排出されず子宮内に貯留(急性腹症で発症も)
-
•
発見時期:思春期の原発性無月経や周期的腹痛で判明することが多い
-
•
推奨:思春期前に腹部超音波やMRIで内性器の評価を実施

🩺 院長コラム【見逃されやすい「隠れたリスク」】
ランガー・ギーディオン症候群について臨床的に重要なのは、女性患者における生殖器異常(ミュラー管形成不全)が見逃されやすいという点です。
出生時や幼児期には気づかれず、思春期になって初経が来ない、あるいは周期的な激しい腹痛で受診して初めて発見されることがあります。子宮留血腫は急性腹症として緊急手術を要することもあります。
私は、LGSと診断された女児に対しては、思春期前の段階で内性器の画像評価を標準ケアに組み込むべきと考えています。早期発見により、閉鎖処女膜の切開などの待機的処置が可能となり、緊急事態を回避できます。
3. 原因と遺伝的背景|3つの責任遺伝子
【結論】 本症候群の原因は、8q24.11-q24.13領域に位置するTRPS1・EXT1・RAD21の3遺伝子のハプロ不全です。これらはそれぞれ骨格形成・外胚葉発生・細胞周期制御に関わる重要な遺伝子であり、一括欠失により複合的な症状が生じます。
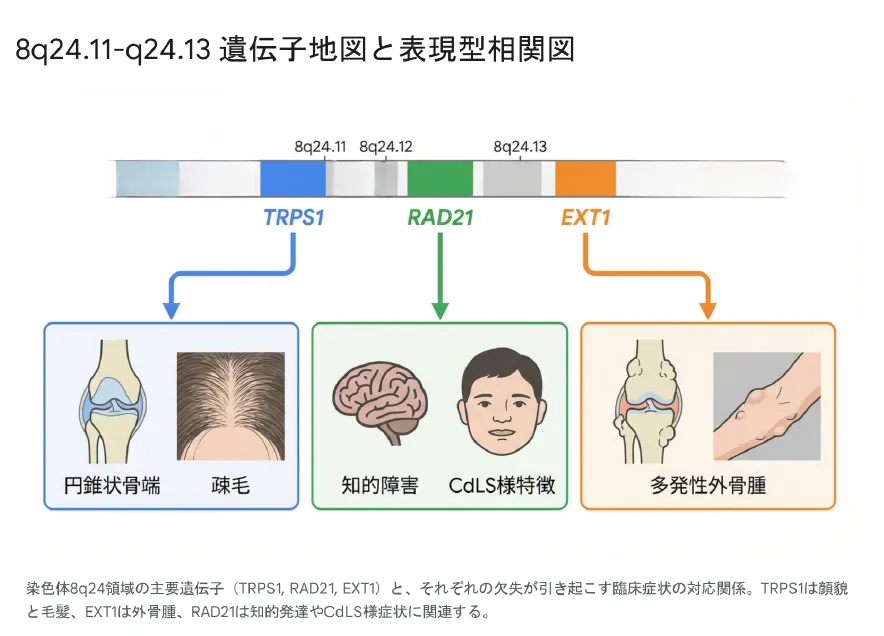
▲ 8q24.11-q24.13領域の主要遺伝子(TRPS1, RAD21, EXT1)と臨床症状の対応関係
💡 用語解説:「ハプロ不全」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。「ハプロ不全」とは、1コピーが欠失または機能しなくなることで、残り1コピーだけでは正常な機能を維持できない状態を指します。遺伝子産物(タンパク質)が通常の50%になることで細胞機能に影響が出ます。
3つの責任遺伝子の機能
| 遺伝子 | 染色体位置 | 主な機能 | 欠失による症状 |
|---|---|---|---|
| TRPS1 | 8q23.3-q24.11 | GATA型転写因子、軟骨細胞の増殖・分化、毛包発生を制御 | 特徴的顔貌、疎毛、円錐状骨端 |
| EXT1 | 8q24.11 | 糖転移酵素、ヘパラン硫酸生合成、腫瘍抑制遺伝子 | 多発性外骨腫 |
| RAD21 | 8q24.11 | コヒーシン複合体構成因子、姉妹染色分体接着、遺伝子発現調節 | 知的障害、成長障害、CdLS様特徴 |
RAD21:コヒーシン病としての側面
💡 用語解説:「コヒーシン病」とは?
コヒーシン複合体(RAD21, SMC1A, SMC3などで構成)は、細胞分裂時に姉妹染色分体を接着させる重要な役割を持ちます。このコヒーシン関連遺伝子の変異による疾患群を「コヒーシン病(Cohesinopathy)」と呼びます。代表的な疾患はコルネリア・デ・ランゲ症候群(CdLS)です。
RAD21の欠失を含むLGS患者では、TRPSの特徴に加えてCdLSに類似した表現型(より重度の知的障害、成長障害、多臓器合併症)が「ブレンド」された状態で現れることがあります。これは、LGSを単なる骨系統疾患としてではなく、コヒーシン病の一種として捉える視点の重要性を示しています。
遺伝形式と発症機序
新生突然変異(大多数)
両親は正常で、配偶子形成時または受精後初期に新たに欠失が発生。家族性の発症は稀であり、ほとんどの症例がde novo(新生突然変異)です。
常染色体顕性(優性)遺伝
欠失を持つ親から子への伝達は50%の確率。ただし、非モザイクの保因者から子への伝達は今のところ報告されていません。
⚠️ 生殖細胞系列モザイクに注意:両親の血液検査で欠失が見つからなくても、生殖細胞系列モザイクの可能性は完全には排除できません。次子への再発リスク評価には慎重な遺伝カウンセリングが求められます。
4. 8q24欠失症候群の診断方法
【結論】 本症候群の確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。臨床症状(3徴)から疑い、CMAで欠失の有無と範囲を特定します。欠失範囲の特定は、RAD21を含むかどうかで予後予測に重要です。
臨床診断基準
以下の3徴(トライアド)が揃っている場合、LGSが強く疑われます。
-
①
外観:疎毛、洋梨状の鼻、薄い上唇
-
②
骨格:多発性外骨腫(触診またはX線)、短指症
-
③
発達:軽度の知的障害または発達遅滞
遺伝学的検査の種類
| 検査方法 | 特徴 | 8q24欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。欠失の正確な範囲・サイズを特定可能 | ◎ 検出可能 |
| G分染法(核型分析) | 数Mb以上の大きな欠失は検出可能 | △ 大きな欠失のみ |
| FISH法 | TRPS1/EXT1領域の特異的プローブで迅速診断 | ○ 検出可能 |
| MLPA法 | 特定領域のコピー数を定量、比較的安価 | ○ 検出可能 |
💡 なぜCMAが重要か?
CMAは欠失の正確な境界とサイズを特定できます。これにより、RAD21が欠失に含まれているかどうかを確認でき、予後予測(知的障害の程度、CdLS様症状の有無)に極めて有用です。G分染法で正常でもLGSが疑われる場合は、必ずCMAを実施すべきです。
5. 治療と生涯にわたる管理
【結論】 本症候群には根本的な治療法は存在せず、症状に応じた対症療法・早期療育・継続的サーベイランスが中心となります。多職種チームによる包括的アプローチが不可欠であり、ライフステージに応じた管理が重要です。
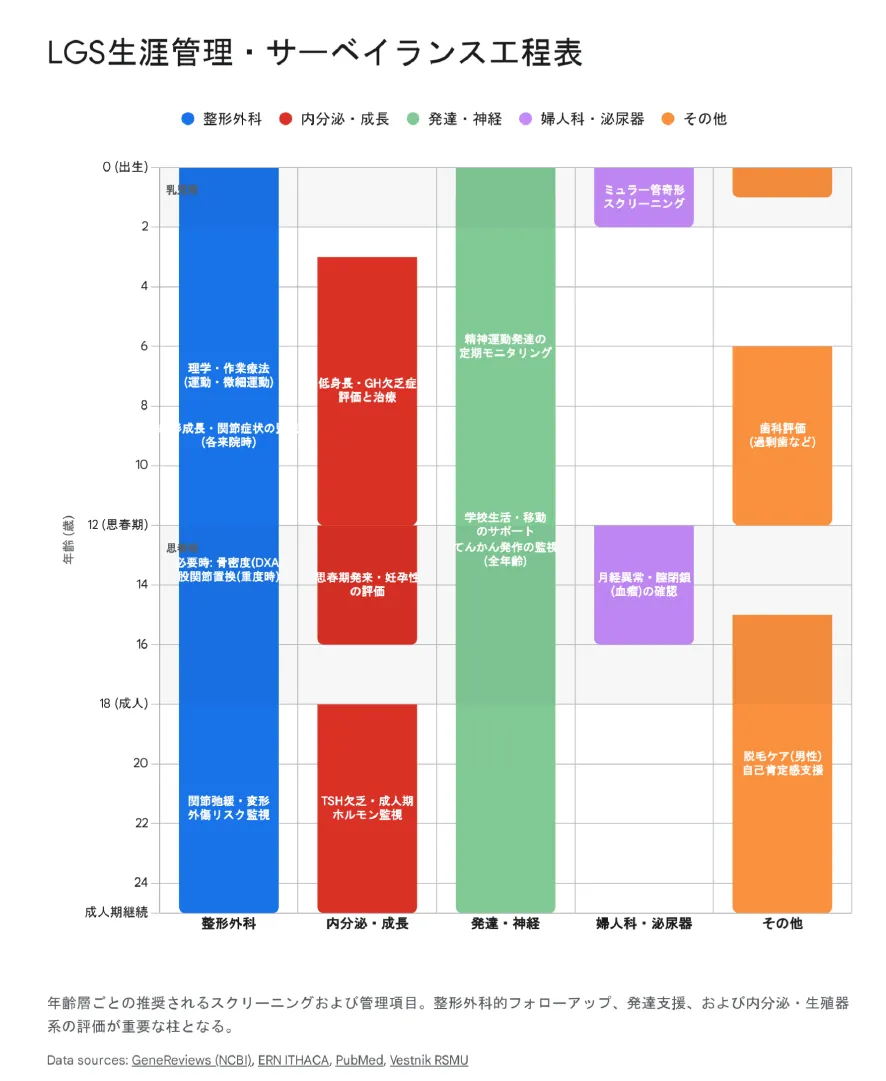
▲ LGS生涯管理・サーベイランス工程表:年齢層ごとの推奨スクリーニングおよび管理項目
ライフステージ別管理指針
| ライフステージ | 主な管理内容 |
|---|---|
| 乳幼児期(0〜5歳) | 成長モニタリング(身長・体重・頭囲)、発達支援(PT・OT・ST)、外骨腫の観察、聴覚・視力スクリーニング、女児は内性器評価 |
| 学童期(6〜12歳) | 整形外科的サーベイランス(年1回以上)、X線での外骨腫・側弯・股関節評価、知能検査、特別支援教育の検討、歯科矯正 |
| 思春期(13〜18歳) | 外骨腫の急速増大に注意、骨端線閉鎖時の全身骨評価、女性は無月経・腹痛に対応、心理的サポート(脱毛・外見への配慮) |
| 成人期(19歳以降) | 変形性関節症への対応、関節弛緩・不安定性の管理、外骨腫の悪性化モニタリング、社会生活・就労支援 |
症状別の治療・対応
外骨腫への対応
- •
全ての外骨腫の切除は不要
- •
疼痛・神経圧迫・可動域制限がある場合に手術検討
- •
急激な増大・疼痛変化は悪性化を疑い精査
- •
接触スポーツは避けることを推奨
発達支援
- •
早期療育が最も重要
- •
理学療法(PT)・作業療法(OT)・言語療法(ST)
- •
特別支援教育の積極的活用
- •
「何ができるか」に焦点を当てた評価
内分泌管理
- •
GH欠乏症があれば成長ホルモン療法
- •
甲状腺機能のスクリーニング
- •
思春期発来遅延への対応
心理社会的支援
- •
脱毛・外見への心理的ケア
- •
ウィッグなど整容的サポート
- •
就労支援・社会的自立促進
予後とQOL
✅ 生命予後は基本的に「正常」
重篤な心疾患や悪性腫瘍の合併がなければ、健常人と同等の寿命が期待できます。ただし、QOLは骨関節症状による慢性疼痛や、外見的特徴に対する心理社会的影響によって制約を受ける可能性があります。適切な医療・福祉介入により自立度と生活の質を最大限に高めることが目標です。
6. 遺伝カウンセリングの重要性
【結論】 8q24欠失症候群と診断された場合、遺伝カウンセリングは不可欠です。診断の意味、予後、再発リスク、そして家族への影響について、正確な情報を提供し、ご家族の意思決定を支援することが重要です。
遺伝カウンセリングで伝えるべきポイント
-
①
欠失範囲と予後の関係:RAD21を含むかどうかで知的障害の程度が異なる
-
②
遺伝形式:常染色体顕性(優性)だが、大多数は新生突然変異
-
③
生命予後:基本的に正常、適切な管理でQOL向上可能
-
④
両親の検査:再発リスク評価のために両親の検査を検討
-
⑤
長期フォローの必要性:多職種チームによる継続的ケア
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常(新生突然変異) | 1%未満(生殖細胞モザイクの可能性はあり) |
| 片親が欠失を保有 | 50% |
🩺 院長コラム【ご家族へのメッセージ】
ランガー・ギーディオン症候群と診断されたとき、ご家族は大きな不安を抱えられることと思います。「この子の将来はどうなるのか」「自分たちにできることは何か」という疑問は当然のことです。
大切なことは、生命予後は基本的に正常であり、適切な医療介入と支援により、多くの患者さんが社会生活を送れているという事実です。また、知的障害が軽度の場合、実用的な生活スキルはIQ以上に良好なことが多いです。
私は臨床遺伝専門医として15年以上のキャリアを持ち、医師としてのべ10万人以上のご家族の意思決定と向き合ってきました。一人で悩まず、ぜひご相談ください。正確な情報を提供し、ご家族と一緒に最善の道を考えていきます。
7. 出生前診断について|NIPTと羊水検査
【結論】 8q24欠失症候群は出生前診断で検出可能です。ミネルバクリニックのNIPTでは8q23q24欠失が検査対象の12 microdeletionsに含まれており、スクリーニングが可能です。確定診断には羊水検査・絨毛検査でのCMAが必要です。
出生前検査での検出方法
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| NIPT(ミネルバ) | ○ スクリーニング可能 | 8q23q24 delが12 microdeletionsに含まれる。COATE法採用 |
| 羊水検査+CMA | ◎ 確定診断可能 | ゴールドスタンダード。欠失範囲も特定可能 |
| 絨毛検査+CMA | ◎ 確定診断可能 | 妊娠初期(11〜14週)に実施可能 |
💡 ミネルバクリニックのNIPTで検出可能な微小欠失症候群(12種)
1p36 del, 2q33 del, 4p16 del, 5p15 del, 8q23q24 del(本症候群), 9p del, 11q23q25 del, 15q11.2-q13 del, 17p11.2 del, 18p del, 18q22q23 del, 22q11.2 del
出生前診断で見つかった場合の対応
-
①
遺伝カウンセリング:疾患の説明、予後、欠失範囲の意味を説明
-
②
両親の検査:親が保因者か確認し、再発リスクを評価
-
③
詳細超音波:骨格異常、心奇形などの構造異常を精査
-
④
出生後フォロー体制:多職種チームによる早期介入の準備
⚠️ 重要:出生前診断で8q24欠失が見つかった場合、生命予後は基本的に正常であることを念頭に置いてください。症状の程度は欠失範囲により異なり、適切な早期介入で良好な経過を辿ることも多いです。どのような決断をされても、専門家としてサポートいたします。
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。8q24欠失症候群を含む染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。8q23q24 delを含む12種の微小欠失をスクリーニング可能です。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会で費用面も安心
互助会(8,000円)により、陽性時の確定検査(羊水検査)費用を全額カバー。上限なしで安心です。
よくある質問(FAQ)
🏥 一人で悩まないでください
8q24欠失症候群(ランガー・ギーディオン症候群)について心配なこと、
検査を受けるかどうか迷っていること、どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
参考文献
- [1] Maas SM, et al. Trichorhinophalangeal Syndrome. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Updated 2017. [GeneReviews]
- [2] Orphanet. Trichorhinophalangeal syndrome type 2. [Orphanet]
- [3] OMIM #150230 – Trichorhinophalangeal Syndrome, Type II; TRPS2. [OMIM]
- [4] Schinzel A, et al. Long-term follow-up of four patients with Langer-Giedion syndrome: clinical course and complications. Am J Med Genet A. 2013;161A(9):2216-2225. [PubMed]
- [5] Delineation of phenotypes and genotypes related to cohesin structural protein RAD21. Hum Genet. 2020;139(5):575-592. [PMC]
- [6] Ludecke HJ, et al. Molecular dissection of a contiguous gene syndrome: localization of the genes involved in the Langer-Giedion syndrome. Hum Mol Genet. 1995;4(1):31-36. [PubMed]
- [7] Genomics Education Programme. Hereditary multiple exostoses. [NHS Genomics Education]
- [8] Favilla BP, et al. Minimal Critical Region and Genes for a Typical Presentation of Langer-Giedion Syndrome. Cytogenet Genome Res. 2022;162(1-2):46-54. [Karger]
- [9] ERN ITHACA. Trichorhinophalangeal Syndrome Clinical Management Guidelines. [ERN ITHACA]
- [10] Chromosome Disorder Outreach. 8q24 Langer-Giedion Syndrome. [CDO]
関連記事







