目次
- 1 【専門医が解説】19番染色体異常(トリソミー・微小欠失)の特徴と予後

【専門医が解説】19番染色体異常(トリソミー・微小欠失)の特徴と予後
📍 クイックナビゲーション
ヒトの19番染色体は、全DNAの約2%という小さなサイズでありながら、ヒトゲノムの中で最も遺伝子密度が高い染色体の一つです。そのため、19番染色体の数の異常(トリソミー等)や構造的異常(微小欠失・微小重複)は、生命の維持や身体の発達に対して極めて重大で多岐にわたる影響を及ぼします。
本記事では、19番染色体の完全トリソミーの致死性メカニズムから、モザイク胚の取り扱い、さらには短腕(19p)や長腕(19q)に発生する微小欠失・微小重複による希少なシンドロームまで、最新の分子遺伝学的知見をもとに臨床遺伝専門医の視点で網羅的かつ詳細に解説します。
1. 19番染色体とは:高密度な遺伝子がもたらす影響
ヒトの19番染色体は、約5,900万個の塩基対で構成されています。サイズとしては小さい部類に入りますが、そこには約1,500個ものタンパク質コード遺伝子が密集して含まれています。この「遺伝子密度の異常な高さ」が、19番染色体の異常を極めて重篤な状態へと導く最大の理由となっています。
遺伝子には、多すぎても少なすぎても正常な機能が損なわれる「遺伝子量効果(Gene dosage effect)」という原則があります。19番染色体においてこのバランスが崩れると、胚の発生、中枢神経の発達、骨格系の成長、そして免疫系や内分泌系に至るまで、生命の根幹に関わる重大なエラーが多発することになります。
💡 遺伝子量効果(Gene dosage effect)とは:
遺伝子から作られるタンパク質の量が、遺伝子のコピー数に比例して増減し、それが細胞や個体の機能に直接影響を与える現象のことです。人間の身体は非常に精巧なバランスで成り立っているため、レシピ(遺伝子)の分量が少しでも狂うと、正常な形が作れなくなってしまいます。
2. 19番染色体完全トリソミーの病態と「致死性」のメカニズム
通常、ヒトの常染色体は父親と母親から1本ずつ受け継ぎ、2本で1対(ペア)となっていますが、これが3本になってしまう状態をトリソミーと呼びます。
13番(パトウ症候群)、18番(エドワーズ症候群)、21番(ダウン症候群)のトリソミーは、重篤な症状を伴いながらも出生に至る可能性があります。しかし、生殖細胞系列(受精卵の段階)における「19番染色体完全トリソミー」が生産児として報告されることはありません。一般的に胚性致死となります。
それどころか、19番トリソミーは自然流産(稽留流産)の組織からさえもほとんど検出されないほど、着床前後の極めて初期の段階で生命維持が困難になるという、桁外れの致死性を持っています。
🔬 なぜ19番トリソミーは着床前後で死滅するのか?(X染色体不活性化の破綻)
19番トリソミーが極めて初期段階で致死となる理由について、最新の分子生物学的研究ではX染色体不活性化(X-chromosome inactivation: XCI)プロセスへの干渉が強く示唆されています。
ヒトの女性は2本のX染色体を持ちますが、胚発生の初期にXIST(Inactive X Specific Transcript)遺伝子の働きにより、1本のX染色体が不活性化され、男女間の遺伝子量が補償されます。
近年の研究により、19番染色体上には、このXISTを抑制し、1本のX染色体を「活性状態」として保護するための「用量依存的なリプレッサー遺伝子(KDM4B遺伝子など)」が存在する可能性が指摘されています。
19番染色体が3本になると、このリプレッサーが過剰に産生され、不活性化プロセスが阻害されてしまいます。
その結果、細胞内に「2本の活性化したX染色体」が存在する状態となり、これが初期胚において決定的な致死性をもたらすと考えられています。この精緻なメカニズムが、胎児が流産として認識される前に消失してしまう理由を分子レベルで説明しています。
3. モザイク型19番染色体トリソミーとPGT-Aにおける課題
すべての細胞がトリソミーである「完全な19番トリソミー」は致死的ですが、近年、体外受精(IVF)における着床前胚染色体異数性検査(PGT-A)の急速な普及に伴い、新たな臨床的課題が浮上しています。
それは、胚盤胞の栄養外胚葉(Trophectoderm: TE細胞、将来胎盤になる部分)から「19番染色体のモザイク」が検出されるケースが日常的に報告されるようになったことです。
💡 モザイク(Mosaicism)とは:
1つの胚(または個体)の中に、正常な染色体を持つ細胞群と、異常な染色体を持つ細胞群が「モザイク画」のように混ざり合って存在している状態を指します。
次世代シーケンシング(NGS)を用いた解析により、30〜50%の異常細胞を含む「低レベルモザイク」と診断された胚を移植した場合、一部は正常な生産児(健康な赤ちゃん)に至る例も確認されています。
なぜ異常細胞が混ざっていたのに健康に生まれるのでしょうか?これは主に以下の2つの理由が推測されます。
- 限局性胎盤モザイク(Confined Placental Mosaicism: CPM): 異常な細胞が、胎児本体ではなく、胎盤などの胚体外組織にのみ限定して存在していた場合。
- 自己修復機能: 胚の発生過程において、正常な細胞が異常な細胞を自然に排除・死滅させ、正常細胞だけが生き残るメカニズムが働いた場合。
しかし、リスクも存在します。極めて稀ではありますが、異常細胞が胎児側に移行して真性モザイクとして生還した症例も報告されています。生還したモザイク症例では、重度の成長障害、好中球減少症、精神運動発達遅滞、および多発奇形などの重篤な合併症が観察されています。
したがって、モザイク胚の移植を検討する際には、慎重な遺伝カウンセリングが必須です。また、妊娠成立後には、超音波検査での厳密な経過観察に加え、妊娠14週以降の羊水穿刺などによる胎児核型の確認(出生前診断)が不可欠なプロセスとなります。
🔍 関連記事:着床前検査と妊娠後の検査について
4. 【特異な二面性】白血病における「後天的な19番トリソミー」の劇的な予後
ここから、医学的に非常に興味深い現象を解説します。生殖細胞系列(受精卵の段階)で発生する19番トリソミーが「致死的」であるのに対し、出生後に大人になってから血液細胞の変異として起こる「後天的な体細胞突然変異としての19番染色体トリソミー」は、全く異なる意味を持ちます。
後天的な19番トリソミーは、血液悪性腫瘍、特に急性骨髄性白血病(AML)や骨髄異形成症候群(MDS)において反復して認められる細胞遺伝学的異常です。MRC(Medical Research Council)の臨床試験データ等に基づく大規模な後方視的解析によると、19番染色体トリソミーは全体の約1%にみられる稀な異常であり、その多く(約82%)は「複雑核型(Complex karyotype)」の一部として現れます。複雑核型の一部として現れる場合の予後は、一般的に不良です。
しかし極めて特筆すべき点として、19番染色体トリソミーを「単独の異常(sole abnormality)」として有するAML患者は、化学療法に対して劇的かつ特異的な反応を示すことが判明しています。
急性骨髄性白血病(AML)における導入療法後の完全寛解率(CR)
AML患者全体
(トリソミー19含む)
単独異常としての
トリソミー19
※MRC(Medical Research Council)の大規模後方視的解析データより作成
上記のグラフが示す通り、統計学的解析において、単独の19番染色体トリソミーを持つ患者は、導入療法後に89%という極めて高い完全寛解(CR)率を示すことが確認されています。これはコホート全体のCR率(52%)を大きく上回っています。
さらに、5年全生存率(OS)および無再発生存率(RFS)の分析においても、単独のトリソミー19は明確に有利な予後因子であることが証明されました。この特定のサブグループにおいては、リスクを伴う同種造血幹細胞移植(allo-HCT)による追加の生存メリットは統計学的に有意ではなく、標準的な強力な化学療法単独で良好なアウトカムが得られる可能性が示唆されています。
同じ染色体のトリソミーであっても、それが「受精卵」に存在する場合は致死となり、「体細胞(血液細胞)」のクローン進化として生じた場合は治療に対する感受性が劇的に高まるという事実は、医学的に全く逆の様相を呈する極めて興味深いモデル論的意義を持っています。
5. 19番染色体短腕(19p)の微小異常:相反する「鏡像の表現型」
近年、マイクロアレイ染色体検査(CMA)や定量的PCRといった技術の進歩により、従来のG分染法などの顕微鏡レベルでは判別不能な微細なDNAの欠失(減少)や重複(増加)が発見されるようになりました。これをコピー数変異(CNV)と呼びます。
💡 コピー数変異(CNV: Copy Number Variation)とは:
ゲノムDNAのある領域が、標準的な2コピー(父母から1つずつ)ではなく、1コピーに減っていたり(微小欠失)、3コピー以上に増えていたり(微小重複)する構造的変化のことです。
19番染色体短腕(19p)の末端領域における微小欠失および微小重複は、欠失と重複で「互いに対照的な表現型(鏡像の表現型)」を示すことが大きな特徴です。
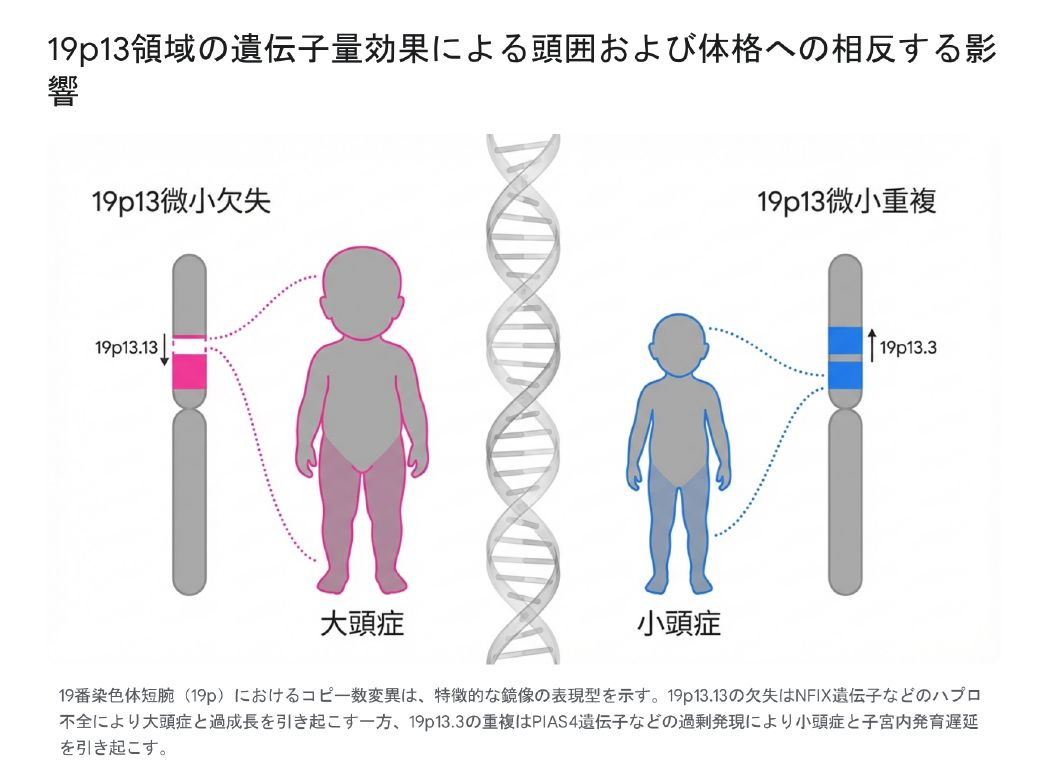
画像:19p13領域の遺伝子量効果による頭囲および体格への相反する影響
5-1. 19p13.13微小欠失症候群(部分モノソミー):大頭症と過成長
19p13.13領域が微細に「欠失」すると、遺伝子の働きが半減するハプロ不全が起こり、身体的な過成長と重篤な神経学的異常を特徴とする稀な症候群を引き起こします。
- 主要な身体的特徴: 最も顕著なのは、異常な頭囲の拡大を伴う大頭症(Macrocephaly)および高身長などの全般的な「過成長(Overgrowth)」です。前頭部の突出や眼瞼裂斜下などの顔面異形態が伴うことも多いです。
- 神経系への甚大な影響: 中等度から重度の知的障害と、発語がほとんどないケースを含む重篤な言語発達遅滞がみられます。運動協調不全(失調)および全般的な筋緊張低下が存在し、座位保持や歩行の著しい遅れに寄与します。また、高頻度で難治性のてんかん発作が合併します。
- 消化器・眼科的異常: 慢性的な便秘、下痢、嘔吐。斜視や、視神経低形成による視力障害の原因となります。
病因となる責任遺伝子と分子メカニズム
この欠失領域には少なくとも16個のタンパク質コード遺伝子が含まれており、これらのハプロ不全が複合的に症状を引き起こします。
- NFIX遺伝子: この遺伝子の欠損が、幼児期の過成長、大頭症、骨格異常を直接的に引き起こします。NFIX単独の変異はMalan症候群の原因でもあり、表現型の強いオーバーラップが分子レベルで裏付けられています。
- CACNA1A遺伝子: カルシウムチャネルをコードし、神経細胞のカルシウムイオン動態異常を引き起こします。てんかん発作や運動失調の直接的な原因です。
- MAST1遺伝子: 神経細胞の分化を阻害し、中枢神経系の発達異常に寄与します。
5-2. 19p13.3微小重複症候群(部分トリソミー):小頭症と免疫不全
微小欠失とは対照的に、19pの末端領域(特に19p13.3)の微小重複は「部分トリソミー」として機能し、遺伝子の過剰発現による全く異なる臨床的特徴をもたらします。
- 身体的特徴: 欠失が大頭症を引き起こすのに対し、重複は子宮内発育遅延(IUGR)、出生後の深刻な低体重、および小頭症(Microcephaly)を引き起こします。この頭囲の逆転現象が明確な遺伝子量効果を示しています。
- 免疫系への甚大な影響: 極めて特異的な症状として、重篤な原発性免疫不全症が挙げられます。IgG1/IgG3の著明な低下、IgM欠乏が確認され、細胞レベルではクラススイッチを経たメモリーB細胞がほぼ完全に消失しています。これが小児期の反復性の重症感染症(肺炎、敗血症など)の原因となります。
- その他の奇形: 重度の精神運動発達遅滞、脳梁低形成。泌尿生殖器奇形(尿道下裂、馬蹄鉄腎)などが報告されています。
病因となる責任遺伝子と分子メカニズム
小頭症および成長障害のメカニズムとして、PIAS4遺伝子の過剰発現が極めて重要な役割を果たしています。PIAS4はSUMO E3リガーゼをコードしており、これが過剰になるとAMPKa1のSUMO化を亢進させ、mTORC1シグナル伝達経路を減弱させます。これにより、胎児期の細胞増殖と頭囲の適切な成長が妨げられるという精緻なメカニズムが解明されています。
表:19p領域における欠失と重複の対比(鏡像の表現型)
| 臨床的・分子的特徴 | 19p13.13 微小欠失(部分モノソミー) | 19p13.3 微小重複(部分トリソミー) |
|---|---|---|
| 頭囲の異常 | 大頭症 (Macrocephaly) | 小頭症 (Microcephaly) |
| 身体的成長 | 過成長、高身長 | 子宮内発育遅延 (IUGR)、低体重 |
| 主要な神経症状 | てんかん(高頻度)、運動失調、筋緊張低下 | 重度の精神運動発達遅滞、脳梁低形成 |
| 特有の合併症 | 消化管機能異常、視神経低形成 | 原発性免疫不全(B細胞異常)、泌尿生殖器奇形 |
| 中核となる責任遺伝子 | CACNA1A, NFIX, MAST1 | PIAS4, MAP2K2, ZBTB7A |
6. 19番染色体長腕(19q)の部分的異常と特徴的な症候群
19番染色体長腕(19q)の構造的異常もまた、特異的で医学的に認識可能な症候群を形成します。ここでは代表的な2つを解説します。
6-1. 19q13.11微小欠失症候群:外胚葉異形成のホールマーク
有病率が100万人に1人未満という極めて稀な疾患ですが、強烈で一貫した症状セットを呈します。
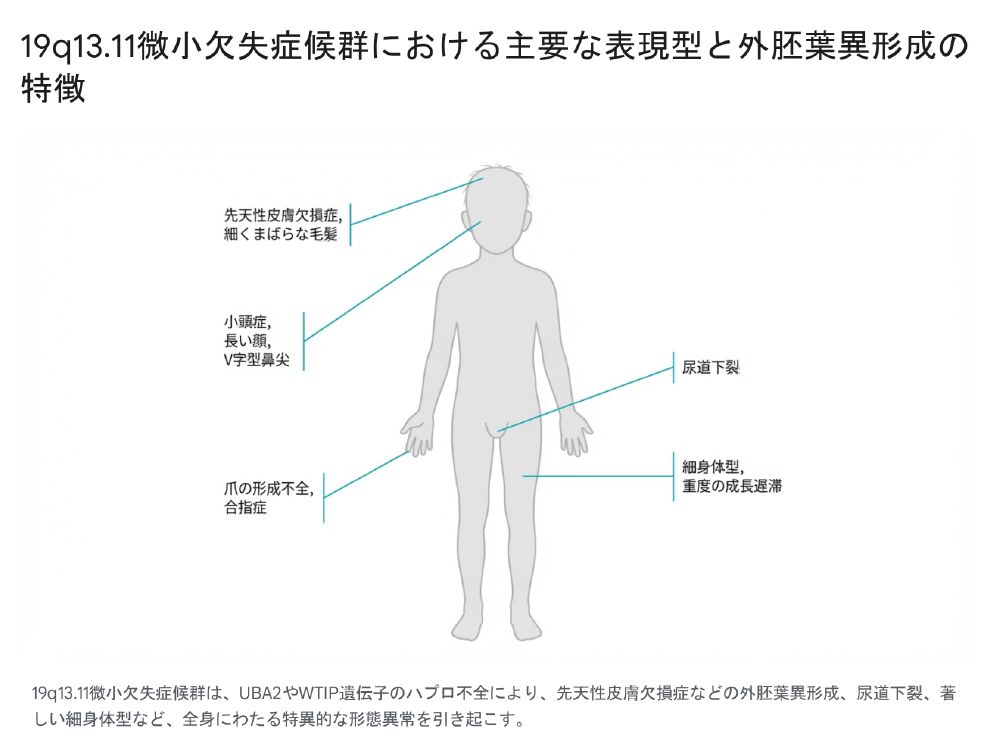
画像:19q13.11微小欠失症候群における主要な表現型と外胚葉異形成の特徴
- 外胚葉異形成(Ectodermal Dysplasia): これが本症候群の最も決定的かつ特異的な特徴(ホールマーク)です。具体的には、正中線上の頭皮が局所的に欠損した状態で出生する先天性皮膚欠損症が高頻度で認められます。また、頭髪・眉毛・まつ毛が極端に細くまばらで、爪の形成不全を伴います。
- 身体・顔貌: 出生前からの重篤な成長遅滞。生後は経管栄養を要するほどの深刻な哺乳困難。皮下脂肪が著しく乏しい細身体型。小頭症、長い顔、V字型の鼻尖。
- 泌尿生殖器奇形: 男性患者ではほぼ全例で尿道下裂や二分陰嚢が観察されます。
- その他: 合指症や裂手裂足(四肢の奇形)を伴うことがあり、EEC症候群と酷似するため鑑別診断が必要です。
💡 外胚葉(がいはいよう)とは:
受精卵が分裂してできる初期の細胞層の一つ。成長するにつれて、皮膚、髪の毛、爪、そして脳や神経系へと分化していきます。この発生過程に異常が生じると、皮膚や毛髪の欠損、神経発達の遅れといった複数の症状がセットで現れます。
病因となる責任遺伝子
- UBA2遺伝子: SUMO活性化酵素のサブユニットをコード。欠損すると外胚葉の発達異常(皮膚欠損や爪の異常)や四肢の奇形(裂手裂足)の直接原因となることがゼブラフィッシュのモデルで示唆されています。
- WTIP遺伝子: 腎臓や生殖器の発生において重要。欠失は男性患者の高頻度な尿道下裂の主要原因遺伝子です。
- KRAB-ZNFクラスター: 欠失領域に密集する転写抑制因子。ハプロ不全が知的障害や神経認知機能の低下に寄与すると考えられています。
6-2. 遠位19q部分トリソミー(重複)
19番染色体長腕の末端領域(19q13.3-qterなど)の重複です。これまで報告された症例の多くは、純粋な突然変異ではなく、親が持つ均衡型転座に由来する不均衡型の染色体分離の結果として生じています。
多臓器にわたる極めて重篤な先天奇形と重度の成長障害を特徴とし、小頭症、平坦な鼻梁、頸部の余剰な皮膚のひだなどの特異な顔貌に加え、先天性心疾患、消化器奇形、難治性のてんかん発作を引き起こします。根本的な治療法はなく、生命維持のための対症療法が中心となるため、予後は不良です。
7. ゲノム構造異常の発生メカニズムと遺伝・再発リスク
なぜDNAの一部が欠けたり(欠失)、増えたり(重複)してしまうのでしょうか。
これは多くの場合、卵子や精子が作られる「減数分裂期」における、染色体間の不均等な交差によって引き起こされます。ゲノム上の似たような配列(低コピー反復配列: LCR)同士が間違って組み合わさってしまう非アレル相同組換え(NAHR)や、DNAが切断された際の修復エラー(非相同末端結合: NHEJ)などが原因で、遺伝子の設計図が物理的に欠落したりコピーされたりします。
💡 常染色体優性(顕性)と劣性(潜性)の考え方:
微小欠失症候群の多くは、2つある遺伝子のうち片方が失われる(ハプロ不全)だけで発症します。これは遺伝学的には「常染色体優性(顕性)の機能喪失」に似た挙動を示します。しかし、親から遺伝したわけではなく、受精卵ができる過程で「突然変異(De novo変異)」として生じるケースが大半です。
お子さんに19番染色体の部分的な異常が発見された場合、臨床管理において極めて重要なのが「ご両親の染色体検査(核型分析)」です。これにより、次のお子さんへの再発リスクが大きく変わります。
遺伝カウンセリングにおける再発リスク評価のステップ
- ① De novo(新生)突然変異の確認: 両親の染色体が正常であれば、今回の異常は生殖細胞形成時などの偶発的なエラーです。次回の妊娠で再発するリスクは一般人口と同等(極めて低い)と見積もられます。
- ② 均衡型転座キャリアの親への対応: 両親のいずれかが、遺伝子の総量に過不足がない「均衡型相互転座」を持っている場合、親自身は健康ですが、減数分裂時に染色体の分離異常が起こりやすくなります。
- ③ リプロダクティブ・オプションの提示: 均衡型転座の場合、不均衡型の配偶子(精子や卵子)ができやすく、不妊や反復流産、染色体異常を持つ子供の出生リスクが高まります。そのため、体外受精と組み合わせた構造異常に対する着床前診断(PGT-SR)や、妊娠成立後の羊水検査などの出生前診断が選択肢となります。
よくある質問(FAQ)
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
関連記事
参考文献
- Consequences of chromosome gain: A new view on trisomy syndromes [PMC]
- Embryonic loss of human females with partial trisomy 19 identifies region critical for the single active X [PMC]
- Impact of trisomy 19 on outcome according to genetic makeup in patients with acute myeloid leukemia [PMC]
- 19p13.13 deletion syndrome – Genetics [MedlinePlus]
- PIAS4 is associated with macro/microcephaly in the novel interstitial 19p13.3 microdeletion/microduplication syndrome [PMC]
- A novel immunodeficiency syndrome associated with partial trisomy 19p13 [PMC]
- 19q13.11 microdeletion concomitant with ins(2;19)(p25.3;q13.1q13.4)dn in a boy: potential role of UBA2 in the associated phenotype [PMC]






