目次

Q. 16p13.3微小欠失症候群とはどのような病気ですか?
A. 16番染色体短腕(16p13.3)にあるCREBBP遺伝子を含む領域が広範囲に欠失する希少疾患です。
Rubinstein-Taybi症候群(RSTS)の特徴に加え、隣接遺伝子(DNASE1等)の欠失により重篤な免疫不全、難治性てんかん、心奇形などを合併することがあり、「重症型RSTS」とも呼ばれます。
-
➤原因 → 16p13.3領域の広範な微小欠失(CREBBP + 隣接遺伝子)
-
➤特徴 → RSTS症状(幅広母指・特徴的顔貌)に加え、致死的な合併症リスクが高い
-
➤診断 → 染色体マイクロアレイ検査(CMA)による欠失範囲の特定が必須
- ➤頻度 → 極めて希少(RSTSの約10%が欠失型、その一部が重症型)
1. 16p13.3微小欠失症候群とは|基本情報
【結論】 16p13.3微小欠失症候群は、16番染色体短腕末端(16p13.3)の微細な領域が欠失することで発症します。この領域にはRubinstein-Taybi症候群(RSTS)の原因遺伝子CREBBPが含まれており、さらに隣接する遺伝子も一緒に欠失することで、より重篤な症状を呈する「連続遺伝子症候群」としての性質を持ちます。

この症候群は、単なるRSTSの一種ではなく、欠失の範囲によって臨床像が大きく異なることが最大の特徴です。典型的RSTSの特徴に加え、生命を脅かす合併症(重症感染症や心奇形)のリスクがあるため、早期の正確な診断と管理が不可欠です。
💡 用語解説:「連続遺伝子症候群」とは?
染色体上の隣り合う複数の遺伝子がまとめて欠失することで、それぞれの遺伝子欠損による症状が合わさり、複雑な病像を呈する疾患群のことです。16p13.3欠失では、RSTSの原因遺伝子だけでなく、免疫や代謝に関わる遺伝子も同時に失われることが問題となります。
16p13.3欠失の分類と概要
| 分類 | 特徴 |
|---|---|
| 古典的RSTS | CREBBP遺伝子単独の変異・小欠失。典型的な顔貌、幅広母指、知的障害。生命予後は比較的良好。 |
| 重症型16p13.3欠失 | CREBBP + 隣接遺伝子(DNASE1など)の広範欠失。重篤な免疫不全、てんかん、心奇形を合併。乳児期死亡リスクあり。 |
| ATR-16症候群 | テロメア側のHBA遺伝子群を含む欠失。αサラセミア(貧血)を合併。 |
2. 遺伝子構成とスペクトラム|欠失範囲が重要
【結論】 欠失がどこまで広がっているかが予後を決定します。CREBBPだけでなく、DNASE1、TRAP1、TBC1D24などの重要遺伝子が含まれるかどうかが、重症化の鍵となります。
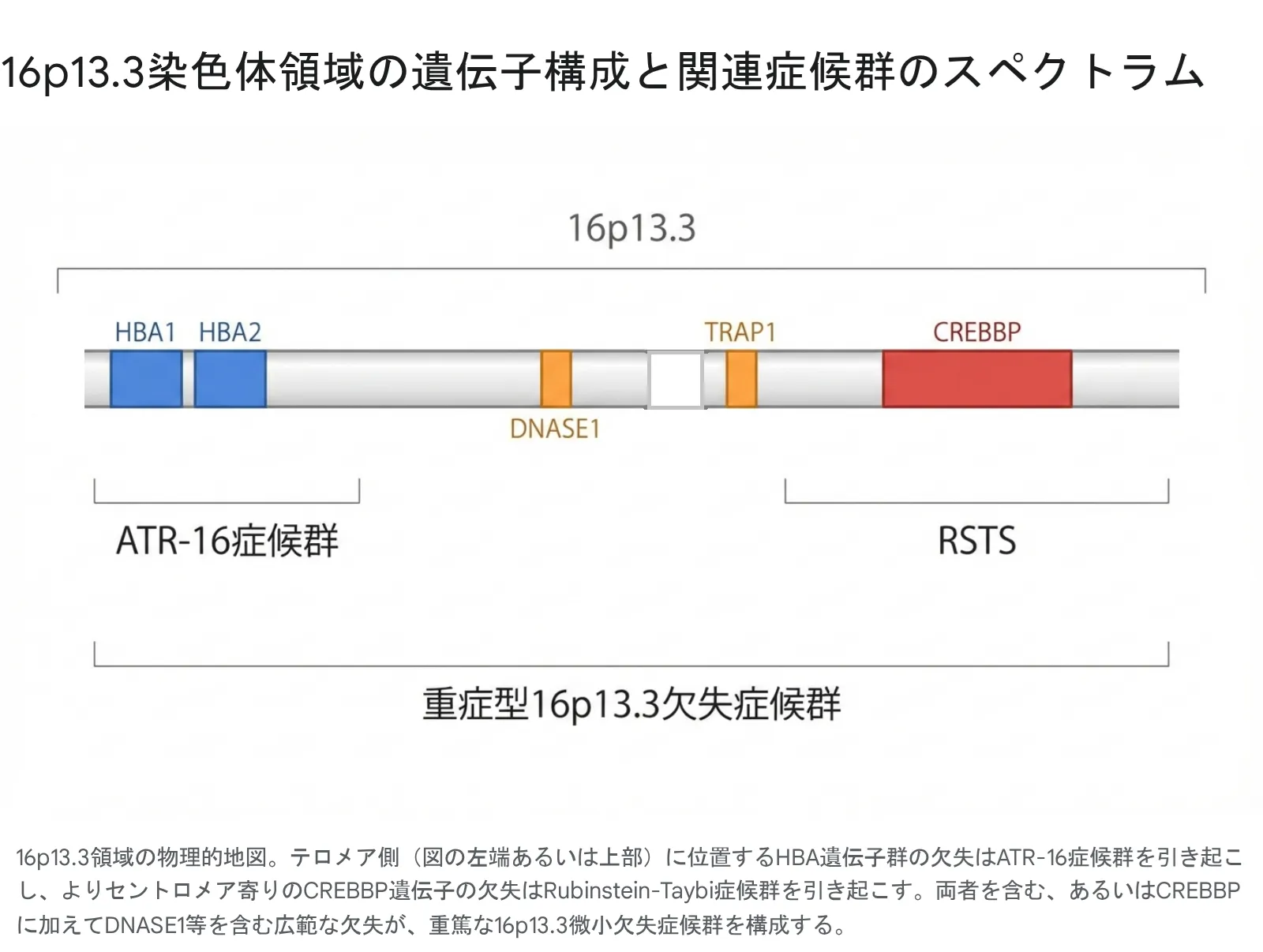
上図のように、16p13.3領域には多数の遺伝子が密集しています。欠失がテロメア側(図の左側)に及ぶとATR-16症候群を合併し、セントロメア側(図の右側)のCREBBP近傍まで拡大すると重症型RSTSとなります。
主な責任遺伝子とその機能
| 遺伝子 | 機能と欠失の影響 |
|---|---|
| CREBBP | 典型的RSTSの主因。転写制御因子。知的障害、骨格異常、顔貌異常を引き起こす。 |
| DNASE1 | 細胞外DNAの分解。欠失により自己免疫疾患(SLE様)や重篤な炎症のリスク増大。 |
| TRAP1 | ミトコンドリアの保護。酸化ストレスへの脆弱性、心筋や神経組織へのダメージに関与。 |
| TBC1D24 | 神経細胞の機能に関与。欠失により難治性てんかんや難聴を引き起こす可能性。 |
| HBA1/HBA2 | αグロビン遺伝子。欠失によりαサラセミア(貧血)を発症(ATR-16症候群)。 |
⚠️ 注意:CREBBP遺伝子単独の変異では免疫不全や重篤な心奇形は稀です。これらが見られる場合は、広範な微小欠失を疑う必要があります。
3. 16p13.3微小欠失症候群の主な症状
【結論】 典型的RSTSの症状(知的障害、幅広母指)に加え、重症型では致死的な感染症、新生児痙攣、多臓器奇形が見られます。以下のマトリックス図で違いを確認してください。
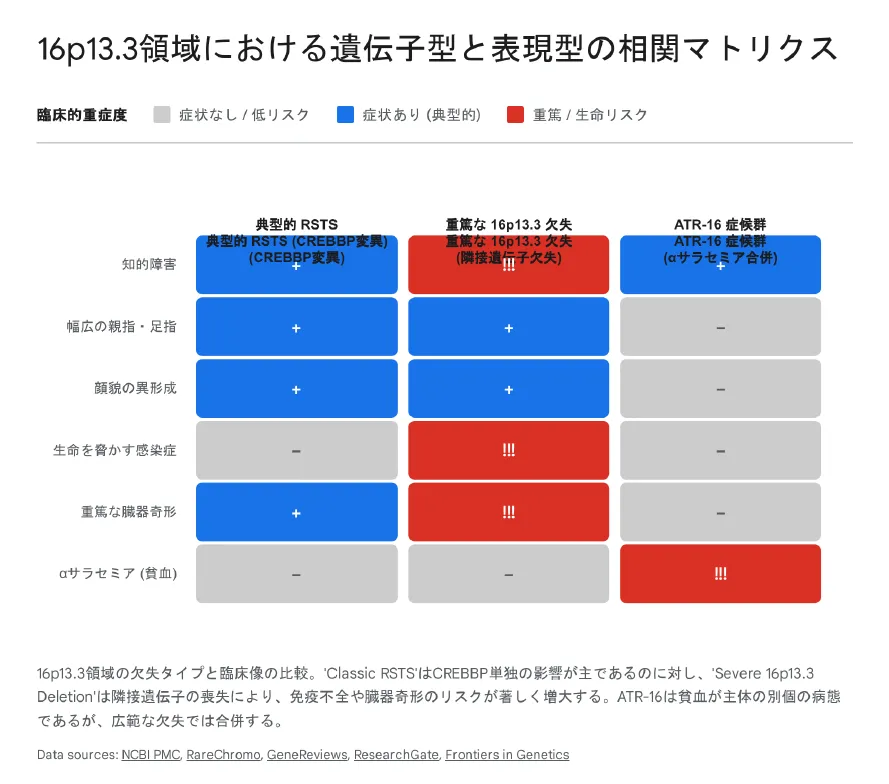
典型的RSTSと共通する症状
-
✓特徴的な顔貌:垂れ目、鷲鼻(ビークノーズ)、口蓋裂、長い睫毛など。
-
✓四肢異常:幅広く短い母指(親指)と母趾(足の親指)。
-
✓発達障害:中等度〜重度の知的障害(IQ 35-50程度)、言語発達遅滞。
重症型16p13.3欠失に特異的な症状
- •
重篤な免疫不全・易感染性:敗血症や髄膜炎など、命に関わる感染症を繰り返す。
- •
難治性てんかん:新生児期からの痙攣発作、コントロール困難なてんかん。
- •
重症心奇形:左心低形成症候群(HLHS)や複雑な血管奇形など。
- •
多臓器奇形:多脾症、内臓逆位、腎奇形(水腎症など)。
4. 診断方法|CMA検査の重要性
【結論】 診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のG分染法では微細な欠失を見逃す可能性があり、FISH法では欠失の範囲(隣接遺伝子が含まれているか)まで特定できないことがあるためです。
診断のステップ
1. 臨床診断
幅広母指、顔貌、発達遅滞の「三徴候」からRSTSを疑います。ただし、新生児期や重症型では特徴が揃わないこともあります。
2. 遺伝学的検査(確定)
CMAで欠失のサイズと場所を特定します。CREBBPだけでなく、DNASE1なども欠失しているかを確認し、重症度や合併症リスクを予測します。
5. 治療とライフステージ別管理
根本的な治療法はないため、合併症の早期発見・治療(サーベイランス)と療育的支援が中心となります。特に新生児期〜乳児期の感染症・心疾患管理が生命予後を左右します。
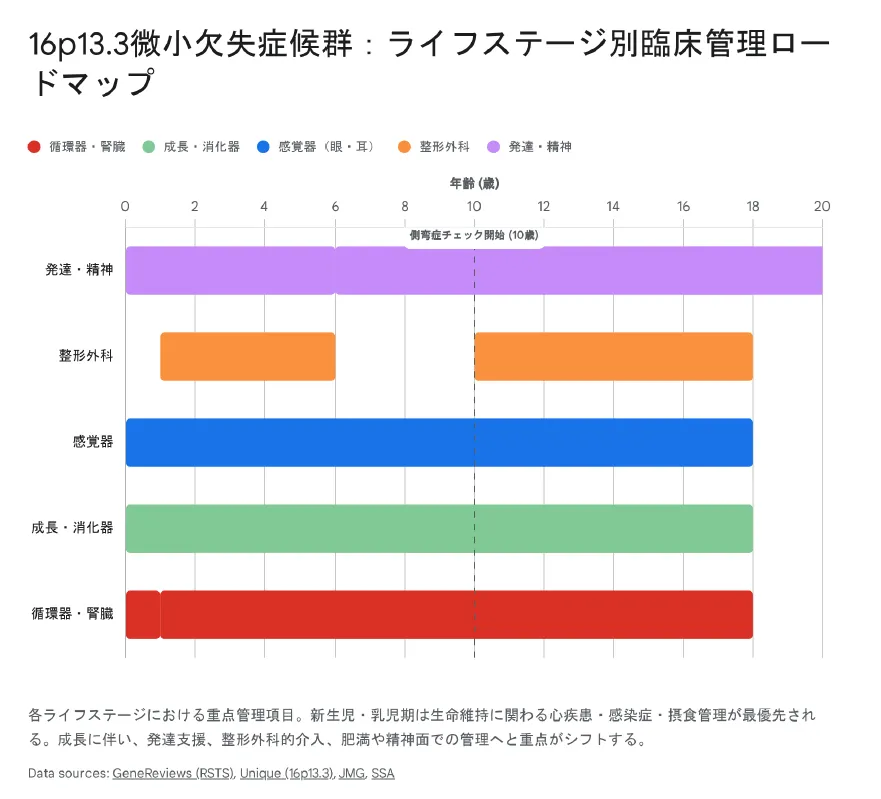
| 時期 | 重点管理項目 |
|---|---|
| 新生児〜乳児期 | 感染症対策(敗血症・髄膜炎)、呼吸管理、哺乳障害への栄養管理、心エコー、新生児痙攣の脳波モニタリング。 |
| 幼児期〜学童期 | 早期療育(PT/OT/ST)、整形外科的介入(母指・母趾の手術)、眼科・耳鼻科検診、肥満予防。 |
| 思春期以降 | 精神心理的ケア、肥満・生活習慣病管理、腫瘍スクリーニング(ケロイド体質に注意)。 |

🩺 院長コラム【RSTSと重症型の違い】
「Rubinstein-Taybi症候群」と診断されても、実はこの「重症型16p13.3欠失」である可能性があります。単なる遺伝子変異と、隣接遺伝子まで含む広範欠失とでは、警戒すべき合併症が全く異なります。
特に免疫不全や難治性てんかんのリスクが高いことを知らずにいると、対応が遅れてしまう恐れがあります。当院ではCMA(マイクロアレイ)等を用いて詳細な遺伝子解析を行い、リスクを正しく評価することを重視しています。
6. 予後と寿命
予後は欠失の範囲と合併症の程度に大きく依存します。
-
➤典型的RSTS:適切な管理下では、成人期までの生存が十分に期待できます。
-
➤重症型16p13.3欠失:重篤な感染症や心疾患により、乳児期死亡のリスクが高くなります。しかし、早期の集中治療管理により生存期間が延長する可能性もあります。
7. 出生前診断について|NIPTと羊水検査
【結論】 16p13.3微小欠失の出生前診断には限界があります。ターゲットとしていないNIPTでは検出されず、全染色体検査であっても7Mb以上の大きな欠失しか検出できません。確定には羊水検査でのマイクロアレイ解析(CMA)が必要ですが、これには倫理的な課題も伴います。
出生前検査での検出方法
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| NIPT(非確定検査) | ✕ 困難 | 全染色体検査でも7Mb以上の欠失しか検出できません。本疾患のような微小欠失は、ターゲットとしていないNIPTでは検出できません。 |
| 羊水検査+CMA | ◎ 検出可能 | 確定診断のゴールドスタンダード。ただし、所見のない胎児への実施は倫理的問題あり。 |
出生前診断で確定させるにはマイクロアレイ検査が必要ですが、超音波検査で所見がない(何もない)胎児に対して侵襲的なマイクロアレイを行うことは、倫理的な問題が含まれます。検査を行うべきかどうかについては、臨床遺伝専門医による十分な遺伝カウンセリングが必要です。
🩺 院長コラム【出生前診断と遺伝カウンセリング】
この疾患は非常に希少で情報も少ないため、出生前診断で見つかるとご家族は大きな不安に包まれます。「どの程度重症なのか?」「生存できるのか?」という問いに対し、遺伝子型に基づいた最新の知見を提供し、ご家族が納得できる意思決定を行えるようサポートするのが私たちの役割です。
当院では、のべ10万人以上のご家族の意思決定と向き合ってきた経験を活かし、中立的かつ温かみのあるカウンセリングを提供しています。
よくある質問(FAQ)
🏥 専門医にご相談ください
16p13.3微小欠失症候群やRSTSの疑い、出生前診断について不安なことがある方は、
ミネルバクリニックの臨床遺伝専門医にご相談ください。
NIPT陽性時の互助会制度(8,000円)による確定検査費用全額補助など、
安心のサポート体制を整えています。
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- [1] Taine L, et al. 16p13.3 microdeletion: a new contiguous gene syndrome. Am J Med Genet. 1998;78(3):267-274. [PubMed]
- [2] Bartsch O, et al. Severe Rubinstein-Taybi syndrome: clinical and molecular studies of 16p13.3 deletions. Eur J Hum Genet. 2006;14(1):17-23. [PubMed]
- [3] Al-Qattan MM, et al. Chromosome 16p13.3 contiguous gene deletion syndrome including the SLX4, DNASE1, TRAP1, and CREBBP genes. Am J Med Genet A. 2020;182(5):1216-1221. [PubMed]
- [4] Kuroda Y, et al. Refinement of 16p13.3 microdeletion syndrome from a case presentation of a girl with epilepsy and intellectual disability. Congenit Anom (Kyoto). 2020;60(6):174-177. [PubMed]
- [5] Mucha BE, et al. 16p13.3 microdeletions: a recognizable syndrome with a distinct phenotype. Genet Med. 2019;21(4):904-912. [PubMed]
- [6] GeneReviews – Rubinstein-Taybi Syndrome. [NCBI]






