目次

Q. 16p11.2-p12.2欠失症候群とはどのような病気ですか?
A. 16番染色体短腕(16p)のp11.2からp12.2にかけての約7〜8Mbという大規模な領域が欠失することで発症する、希少な隣接遺伝子症候群です。
近位16p11.2微小欠失症候群(約600kb)とは明確に区別される独立した疾患であり、平坦な顔貌・重篤な言語遅滞・中等度〜重度の知的障害・反復性中耳炎などを特徴とします。
-
➤
原因 → 16番染色体BP1-BP2間(約7〜8Mb)の大規模欠失、ほとんどが新生突然変異 -
➤
主要症状 → 平坦な顔貌、重篤な言語遅滞、中等度〜重度知的障害、反復性中耳炎、摂食困難 -
➤
重要な特徴 → 近位16p11.2欠失(約600kb)とは異なる独立した症候群、より重篤な臨床像 -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
頻度 → 極めて稀(1/1,000,000未満)、2007年に初めて独立した症候群として報告
1. 16p11.2-p12.2欠失症候群とは|基本情報
【結論】 16p11.2-p12.2欠失症候群は、16番染色体短腕のp11.2からp12.2領域にかけての約7〜8Mbが欠失することで発症する、極めて稀な隣接遺伝子症候群です。2007年にBallifらによって独立した新しい微小欠失症候群として初めて報告されました。
「お子さんの発達が気になる」「検査で16p11.2-p12.2欠失が見つかった」という方は、この病気について正確な情報を知ることが大切です。本症候群は近位16p11.2微小欠失症候群(約600kb)とは異なる独立した疾患であり、より重篤な臨床像を呈することが特徴です。

16番染色体の全体像。16p11.2-p12.2欠失症候群は短腕(p)の広範な領域に及ぶ大規模欠失です。
💡 用語解説:「隣接遺伝子症候群」とは?
隣接遺伝子症候群(contiguous gene syndrome)とは、染色体上で隣接して並ぶ複数の遺伝子がまとめて欠失または重複することで発症する疾患群です。16p11.2-p12.2欠失では約7〜8Mbという広大な領域に含まれる多数の遺伝子のハプロ不全が複合的な症状を引き起こします。
16p11.2-p12.2欠失症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 16p11.2-p12.2欠失症候群(16p12.2-p11.2 deletion syndrome) |
| OMIM | Chromosome 16p12.2-p11.2 deletion syndrome, 7.1- to 8.7-Mb |
| 欠失サイズ | 約7.1〜8.7Mb(近位16p11.2欠失の約13倍) |
| 頻度 | 極めて稀(1/1,000,000未満と推定) |
| 遺伝形式 | 常染色体優性(顕性)、ほとんどが新生突然変異 |
| 主要な候補遺伝子 | SH2B1、KCTD13、TBX6、MAPK3など多数 |
⚠️ 近位16p11.2微小欠失症候群との違い
本症候群は、自閉症スペクトラム障害(ASD)や肥満との関連でよく知られる「近位16p11.2微小欠失症候群(約600kb)」とは明確に異なる疾患です。16p11.2-p12.2欠失は近位欠失の約13倍もの大きさがあり、より多くの遺伝子を含むため、顔貌の異形、重篤な言語遅滞、反復性中耳炎など特徴的な「症候群的表現型」を呈します。
近位16p11.2欠失との比較
臨床現場において最も重要なのは、この2つの異なる症候群を正確に区別することです。以下の表で主な違いを整理します。
| 特徴 | 近位16p11.2欠失(約600kb) | 16p11.2-p12.2欠失(約8Mb) |
|---|---|---|
| 主な神経症状 | 軽度ID、学習障害、ASDが主 | 中等度〜重度ID、重篤な言語遅滞 |
| 顔貌の特徴 | 非特異的、あるいは軽微 | 平坦な顔貌、眼瞼裂斜下、耳介変形など明瞭な異形 |
| 頭囲 | 大頭症(Macrocephaly)傾向 | 小頭症または正常、大頭症は稀 |
| 成長・体格 | 肥満(小児期以降に顕著) | 低身長、乳児期摂食障害、痩せ〜肥満と多様 |
| 特徴的な合併症 | 脊椎異常、てんかん(約25%) | 反復性中耳炎、心疾患、腎奇形、脊椎異常 |
| 行動特性 | ASDが中核的特徴 | 多動・注意欠陥が顕著(ASDは必須ではない) |
💡 なぜ区別が重要なのか?
16p11.2-p12.2欠失は、単なる「近位欠失の拡大版」ではありません。顔貌の異形、反復性中耳炎、摂食障害といった付加的な「症候群的特徴」を持つ、より重篤で独立した疾患です。正確な診断は適切な管理計画の立案に不可欠です。
2. 16p11.2-p12.2欠失症候群の主な症状
【結論】 本症候群は平坦な顔貌、重篤な言語発達遅滞、中等度〜重度の知的障害、反復性中耳炎を中核症状とします。Battagliaら(2009)やHempelら(2009)の詳細な症例報告により、認識可能な症候群パターンが確立されています。
頭蓋顔面の特徴(Craniofacial Dysmorphology)
本症候群の最も視覚的に認識可能な特徴の一つは、特有の顔貌です。文献では「平坦な顔貌(Flat facies)」という表現が繰り返し用いられています。
-
•
全体像:平坦な顔貌(中顔面低形成)、横顔で鼻根部が低い
-
•
眼部:眼瞼裂斜下、眼間開離、深い眼窩、眼瞼下垂
-
•
耳部:耳介低位・後方回転、耳介の形態異常
-
•
口部・顎:高口蓋、口蓋裂または口唇裂、小顎症、テント状の上口唇
神経発達の特徴
-
•
知的障害:中等度〜重度(IQ 50未満となるケースも珍しくない)
-
•
言語発達:発語の著しい遅れまたは欠如(3〜4歳で有意味語なしの例も)
-
•
特徴:表出性言語(話すこと)の障害が受容性言語(理解すること)より重い
-
•
運動面:全身的な筋緊張低下、運動発達遅滞(定頸、座位、独歩の遅延)
行動・精神医学的特性
興味深いことに、本症候群では近位16p11.2欠失ほどASDが中核的ではありません。Battagliaらの報告では、患児は多動で集中力が乏しいものの、形式的な検査ではASDの診断基準を満たさないケースが記述されています。
多動・注意欠陥
- •
多動性が高頻度
- •
注意散漫、衝動性
- •
ADHD様症状
自閉症との関係
- •
ASDの合併例もあるが必須ではない
- •
近位欠失よりASD頻度は低い
- •
発達遅滞・身体異常が主体
全身性の合併症
| 臓器系 | 合併症 | 備考 |
|---|---|---|
| 耳鼻咽喉 | 反復性中耳炎 | 本症候群の特徴的所見、伝音性難聴の原因に |
| 心血管 | 先天性心疾患(17〜30%) | 心房中隔欠損、心室中隔欠損、肺動脈弁狭窄 |
| 消化器 | 摂食障害、まれにヒルシュスプルング病 | 乳児期の経管栄養が必要な例も |
| 泌尿器 | 腎奇形、停留精巣 | 腎欠損、重複腎盂尿管など |
| 骨格 | 脊椎異常、側弯症、長い指 | TBX6遺伝子の関与が示唆される |
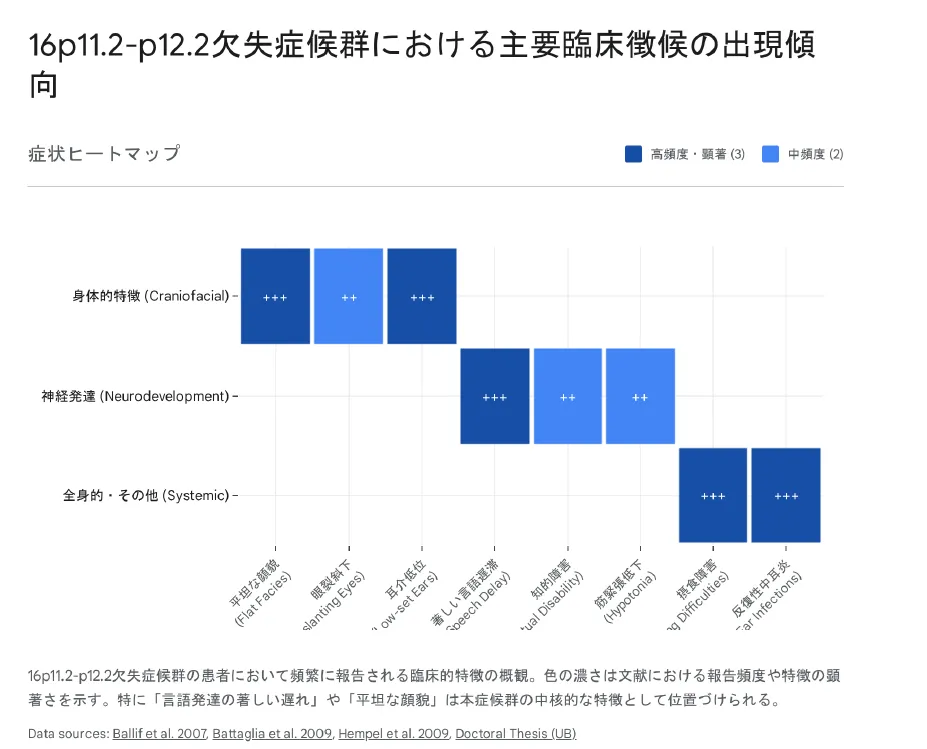
16p11.2-p12.2欠失症候群における主要臨床徴候の出現傾向。平坦な顔貌、言語発達の著しい遅れ、摂食困難、反復性耳炎が高頻度で報告されています。

🩺 院長コラム【症状の個人差について】
16p11.2-p12.2欠失症候群の臨床像は、患者さんごとに大きな幅があることを知っておいてください。報告された症例数がまだ少ないため(2012年までに6例程度)、症状の全体像は完全には解明されていません。
ただし、「平坦な顔貌」「重篤な言語遅滞」「反復性中耳炎」という3つの特徴は、この症候群を近位16p11.2欠失から区別する重要な手がかりとなります。お子さんにこれらの特徴が見られ、原因が不明な場合は、臨床遺伝専門医にご相談ください。
3. 原因と遺伝的背景|発生機序と候補遺伝子
【結論】 本症候群は、16番染色体短腕の分節重複(Segmental Duplications)領域における非対立遺伝子間相同組換え(NAHR)によって発生します。欠失領域には神経発達、代謝調節、骨格形成に重要なSH2B1、KCTD13、TBX6、MAPK3など多数の遺伝子が含まれます。
発生機序:NAHRによる欠失
💡 用語解説:「NAHR」とは?
NAHR(Non-Allelic Homologous Recombination:非対立遺伝子間相同組換え)は、染色体上に散在する高度に類似した反復配列(LCR/SD)同士が誤って対合・組換えを起こすことで、間の領域が欠失または重複する現象です。16番染色体短腕はこの反復配列が特に多く、構造的に不安定な領域として知られています。
16p11.2-p12.2領域には、互いに98%以上の相同性を持つ分節重複(SD)クラスターが複数存在します。減数分裂時にこれらのSD間でNAHRが生じると、約7〜8Mbにわたる遺伝物質が失われます。
新生突然変異(大多数)
両親は正常で、配偶子形成時または受精後初期に新たに発生。本症候群のほとんどは新生突然変異として起こります。次子への再発リスクは1%未満です。
遺伝(稀)
稀に、親が均衡型転座保因者やモザイク変異を有している場合があります。この場合、次子にも欠失が生じるリスクがあるため、両親の検査が重要です。
主要な候補遺伝子とその機能
広範な欠失領域内には多数の遺伝子が含まれており、それぞれが本症候群の多様な症状に寄与していると考えられています。
| 遺伝子 | 主な機能 | 関連する臨床症状 |
|---|---|---|
| SH2B1 | インスリン・レプチンシグナル調節 | 肥満(小児期以降)、インスリン抵抗性 |
| KCTD13 | 細胞骨格制御、頭部サイズ調節 | 頭囲異常(欠失で大頭症傾向、他の遺伝子との相互作用) |
| TBX6 | 中胚葉分化、体節形成 | 脊椎異常、先天性側弯症、半椎 |
| MAPK3 | シナプス可塑性、脳発達 | 知的障害、学習・記憶の問題 |
| OTOA | 内耳機能 | 難聴・反復性中耳炎との関連が示唆 |
💡 用語解説:「ハプロ不全」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。「ハプロ不全(haploinsufficiency)」とは、1コピーが欠失することで、残り1コピーだけでは正常な機能を維持できない状態を指します。本症候群では多数の遺伝子が同時にハプロ不全となるため、複合的な症状が生じます。
🎯 相加的・相乗的効果
重要な点として、16p11.2-p12.2欠失は複数の独立した臨床的意義を持つ小領域を包括しています:
① 近位16p11.2領域(BP4-BP5):自閉症、統合失調症、肥満、大頭症と関連
② 遠位16p11.2領域(BP2-BP3):SH2B1を含み、肥満や行動異常と関連
③ 16p12.1領域:発達遅滞、神経心理学的問題への感受性
本症候群の表現型は、これら個々の領域の効果が「相加的」または「相乗的」に作用した結果として理解されます。
4. 16p11.2-p12.2欠失症候群の診断方法
【結論】 本症候群の確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。約7〜8Mbの欠失は従来のG分染法でも検出可能な場合がありますが、正確なブレイクポイントの特定と近位16p11.2欠失との鑑別にはCMAが必須です。
検査を考慮すべき臨床像
-
①
原因不明の中等度〜重度発達遅滞・知的障害
-
②
重篤な言語表出の遅れ(年齢不相応な無発語など)
-
③
特徴的な顔貌(平坦な顔、耳介低位など)
-
④
乳児期摂食困難と反復性中耳炎の既往
-
⑤
多発奇形(心疾患、骨格異常など)の合併
遺伝学的検査の種類
| 検査方法 | 特徴 | 16p11.2-p12.2欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。正確なブレイクポイントと含まれる遺伝子を同定 | ◎ 検出可能(推奨) |
| G分染法(核型分析) | 解像度は5〜10Mb程度。約8Mbの欠失は検出可能な場合も | △ 検出可能だが詳細は不明 |
| FISH法 | 特定領域のプローブを使用。家族検査に有用 | ○ ターゲットを絞って検出可能 |
💡 用語解説:染色体マイクロアレイ(CMA)とは?
CMAは、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。16p11.2-p12.2欠失の正確な範囲を決定し、近位16p11.2欠失との鑑別において決定的な役割を果たします。日本では原因不明の発達遅滞・先天異常に対する保険適用検査として実施されています。
合併症の評価
診断が確定した場合、以下の臓器合併症スクリーニングが推奨されます。
-
•
心エコー検査:先天性心疾患(ASD、VSD等)のスクリーニング
-
•
腎超音波検査:腎奇形の検索
-
•
聴覚スクリーニング:難聴(OTOA遺伝子関連)および中耳炎の評価
-
•
脊椎画像検査:椎骨異常の確認
-
•
脳MRI:臨床的に示唆される場合(Chiari奇形など)
5. 治療と長期管理
【結論】 本症候群には根本的な治療法は存在せず、症状に応じた対症療法・早期療育・継続的支援が中心となります。多職種チームによる包括的アプローチと、ライフステージに応じた長期的なサーベイランスが重要です。
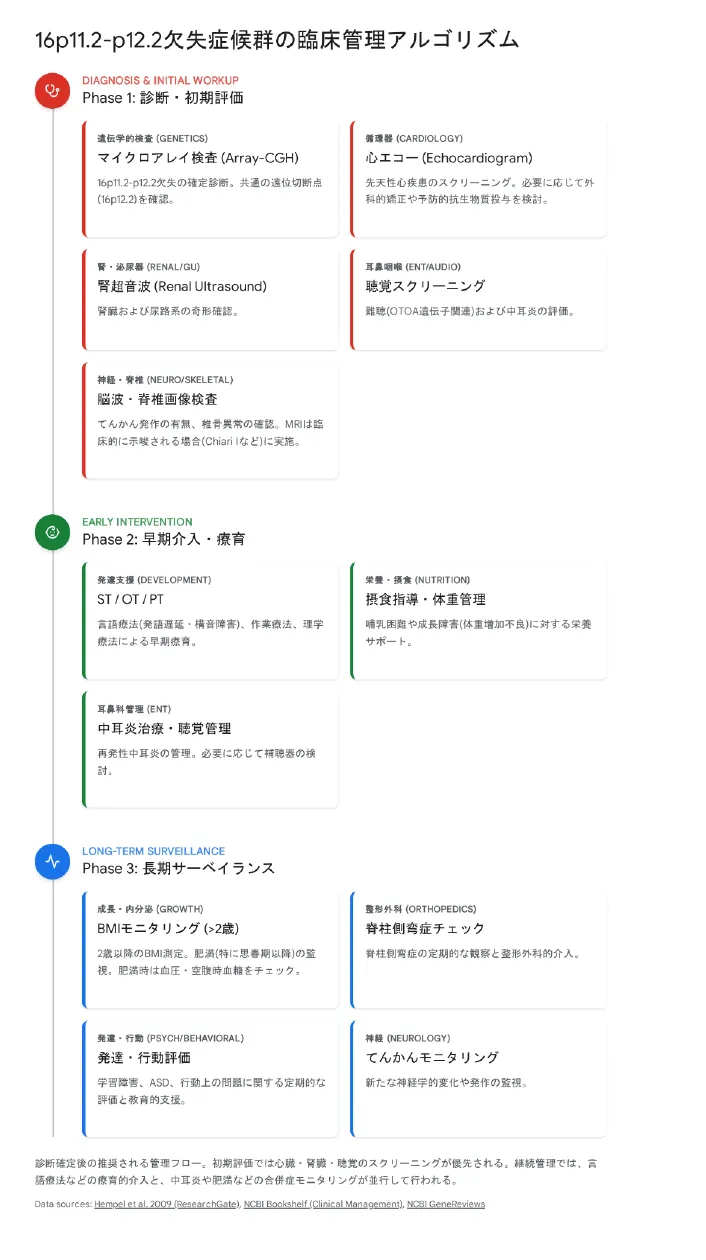
16p11.2-p12.2欠失症候群の臨床管理アルゴリズム。診断・初期評価 → 早期介入・療育 → 長期サーベイランスの3段階で管理します。
ライフステージ別の管理
| ライフステージ | 主な対応 |
|---|---|
| 乳児期(0〜1歳) | 摂食支援(経管栄養の検討)、心エコー、腎超音波、聴覚スクリーニング |
| 幼児期(1〜5歳) | 早期療育開始(PT・OT・ST)、中耳炎の管理、発達評価 |
| 学童期(6〜12歳) | 特別支援教育の検討、ADHD・行動問題への対応、肥満モニタリング |
| 思春期・成人期(13歳〜) | 側弯症チェック、肥満関連疾患の管理、就労・生活支援、移行期医療 |
症状別の治療・対応
発達遅滞・言語障害
- •
早期療育が最重要
- •
言語聴覚療法(ST)を集中的に
- •
AAC(拡大代替コミュニケーション)の導入
- •
特別支援教育の利用
反復性中耳炎
- •
抗生剤治療
- •
鼓膜換気チューブ留置術の検討
- •
難聴があれば補聴器を評価
先天性心疾患
- •
小児心臓専門医との連携
- •
必要に応じて手術・カテーテル治療
- •
定期的な経過観察
摂食・栄養
- •
乳児期:経管栄養の検討
- •
幼児期以降:肥満予防の栄養指導
- •
定期的なBMI測定
長期予後
本症候群の長期的な自然歴に関するデータはまだ蓄積途上ですが、既存の症例報告からは以下のことが示唆されています。
📊 予後に関する知見
・言語発達:早期療育により一定の改善が期待できる(「2歳で単語が出ず、6歳頃から文で話せるようになった」例の報告)
・知的障害:生涯にわたり継続的支援が必要となるケースが多い
・生命予後:重篤な心疾患や腎疾患の合併がなければ、比較的良好と考えられる
・成人期の課題:肥満関連疾患(糖尿病、心血管疾患)の管理が健康寿命を左右する鍵
6. 遺伝カウンセリングの重要性
【結論】 16p11.2-p12.2欠失症候群と診断された場合、遺伝カウンセリングを通じて疾患の遺伝形式、再発リスク、長期的なフォローアップ体制について丁寧に説明することが重要です。ほとんどが新生突然変異であり、次子への再発リスクは低いことを伝えます。
遺伝カウンセリングで伝えるべきポイント
-
①
発生様式:大多数は新生突然変異(両親は正常)
-
②
両親の検査:稀に均衡型転座保因者やモザイクの可能性あり
-
③
次子への再発リスク:新生突然変異の場合は1%未満
-
④
患者本人が挙児する場合:50%の確率で遺伝(常染色体優性(顕性))
-
⑤
長期フォロー:多職種チームによる継続的サポート体制
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常(新生突然変異) | 1%未満(生殖細胞モザイクの可能性はあり) |
| 片親が均衡型転座保因者 | 上昇(具体的リスクは転座の種類による) |
| 患者本人が挙児する場合 | 50%(常染色体優性(顕性)遺伝) |
🩺 院長コラム【遺伝カウンセリングで大切にしていること】
16p11.2-p12.2欠失症候群は2007年に初めて報告された比較的新しい疾患概念であり、報告症例数もまだ少ないため、「予後の幅が大きい」という不確実性が存在します。
遺伝カウンセリングでは、この不確実性を正直にお伝えしつつも、「早期療育で改善が期待できる部分もあること」「多職種チームで長期的にサポートする体制があること」を丁寧にご説明しています。
私は臨床遺伝専門医として、15年の実績、医師としてのべ10万人以上のご家族の意思決定と向き合ってきました。どのような決断をされても、その後もサポートを続けることをお約束しています。不安を抱えている方は、ぜひ一度ご相談ください。
7. 出生前診断について
【結論】 16p11.2-p12.2欠失は出生前診断で検出可能です。羊水検査・絨毛検査でのCMAが確定診断に有効です。約8Mbという大きなサイズのため、一部のNIPTでも検出される可能性がありますが、ミネルバクリニックの標準パネルには含まれていません。
出生前検査での検出
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| NIPT(スクリーニング) | △ 一部で検出可能 | 約8Mbのため検出される場合もあるが、ミネルバの標準パネル(12種)には含まれない |
| 羊水検査+CMA | ◎ 検出可能 | 確定診断のゴールドスタンダード |
| 絨毛検査+CMA | ◎ 検出可能 | 妊娠初期(11〜14週)に実施可能 |
⚠️ ミネルバクリニックのNIPTで検出可能な微小欠失について
ミネルバクリニックのNIPTで検出可能な微小欠失は以下の12種類です:
1p36欠失、2q33欠失、4p16欠失(Wolf-Hirschhorn)、5p15欠失(Cri-du-chat)、8q23q24欠失、9p欠失、11q23q25欠失(Jacobsen)、15q11.2-q13欠失(PWS/AS領域)、17p11.2欠失(Smith-Magenis)、18p欠失、18q22q23欠失、22q11.2欠失(DiGeorge)
16p11.2-p12.2欠失はこのパネルには含まれていません。ただし、COATE法は全染色体検査には対応していません。16番染色体の異数性自体は検査できます。
出生前診断で見つかった場合の対応
出生前にこの欠失が見つかった場合、超音波検査で明らかな異常所見がないこともあります。予後予測は困難ですが、以下の対応が推奨されます。
-
①
遺伝カウンセリング:欠失の意味、症状の幅、予後の不確実性を説明
-
②
両親の検査:均衡型転座の有無を確認(再発リスク評価)
-
③
詳細超音波:心奇形、口蓋裂などの構造異常を精査
-
④
出生後フォロー体制:発達モニタリング、早期療育の準備
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。16p11.2-p12.2欠失症候群を含む染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🔬 高精度な検査技術
スーパーNIPT(第3世代)とCOATE法を採用。全染色体検査や微小欠失検査も対応可能です。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会制度で費用面も安心
互助会(8,000円)により、陽性時の確定検査(羊水検査)費用が全額カバーされます。上限なしで安心です。
よくある質問(FAQ)
🏥 一人で悩まないでください
16p11.2-p12.2欠失症候群について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- Ballif BC, et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat Genet. 2007;39(9):1071-1073. PubMed
- Hempel M, et al. Microdeletion syndrome 16p11.2-p12.2: clinical and molecular characterization. Am J Med Genet A. 2009;149A(10):2106-2112. PubMed
- Battaglia A, et al. Further characterization of the new microdeletion syndrome of 16p11.2-p12.2. Am J Med Genet A. 2009;149A(6):1200-1204. PubMed
- Tabet AC, et al. 16p11.2-p12.2 duplication syndrome; a genomic condition differentiated from euchromatic variation of 16p11.2. Eur J Med Genet. 2012;55(12):600-604. PMC
- Zufferey F, et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 2012;49(10):660-668.
- 16p11.2 Recurrent Deletion – GeneReviews® – NCBI Bookshelf. NCBI Bookshelf
- 16p12.2 Recurrent Deletion – GeneReviews® – NCBI Bookshelf. NCBI Bookshelf
- 16p11.2p12.2 microdeletion syndrome – Orphanet. Orphanet
- 16p11.2-p12.2 microdeletion syndrome – DECIPHER. DECIPHER
- 16p11.2 deletion syndrome – Wikipedia. Wikipedia
- Unique – Rare Chromosome Disorder Support Group. 16p11.2 microdeletions. RareChromo.org
- Simons Searchlight. 16p11.2 Deletion Guidebook. Simons Searchlight






