目次


📍 クイックナビゲーション
1q41-q42欠失症候群は、第1染色体長腕(1q41-q42領域)の一部が失われることで発症する、極めて稀少な染色体微小欠失症候群です。重度の発達遅滞・知的障害・てんかん・特徴的な顔貌に加えて、先天性横隔膜ヘルニア(CDH)の合併が新生児期の生命予後を大きく左右するという特徴を持ちます。
従来のGバンド染色体検査では捉えきれない微小な欠失であるため、染色体マイクロアレイ検査(CMA)の臨床導入に伴って独立した症候群として確立されました。欠失領域内のWDR26・FBXO28・DISP1・HLXなど複数の遺伝子が同時に失われる「隣接遺伝子症候群」であり、欠失範囲によって症状の重症度や臓器障害のパターンが大きく異なります。
本記事では、最新の分子遺伝学的知見と臨床データをもとに、1q41-q42欠失症候群の原因・症状・診断・治療・予後・遺伝カウンセリング・出生前診断の各論点を、臨床遺伝専門医の視点から網羅的に解説します。
1. 1q41-q42欠失症候群とは|疾患の基本情報
1q41-q42欠失症候群は、第1染色体長腕の1q41からq42領域(約1.17〜6Mb程度)が部分的に失われることで発症する、極めて稀少な染色体微小欠失症候群です。重度の発達遅滞・知的障害・てんかん・特徴的な顔貌・先天性横隔膜ヘルニアなどを主症状とし、欠失領域内の複数の遺伝子が同時に失われることで多臓器に影響が現れる「隣接遺伝子症候群(contiguous gene syndrome)」に分類されます。
表現型のスペクトラムは非常に幅広く、新生児期に致死的な経過をたどる重症例から、軽度の発達遅滞にとどまり成人期まで生活される方まで、患者さんごとに大きな違いがあります。世界全体での報告例は数十例規模にとどまる希少疾患ですが、染色体マイクロアレイ検査の普及により、診断例は徐々に増加しています。
染色体上で隣り合って並んでいる複数の遺伝子が一度に失われることで起こる病気の総称です。それぞれの遺伝子が異なる役割を担っているため、脳・心臓・骨格・呼吸器など複数の臓器に同時に影響が出るのが特徴です。22q11.2欠失症候群やプラダー・ウィリ症候群なども、このグループに含まれます。
1.1 疾患の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 1q41-q42欠失症候群(OMIM #612530) |
| 英語表記 | 1q41q42 microdeletion syndrome |
| 原因 | 第1染色体長腕(1q41-q42領域)の微小欠失 |
| 頻度 | 極めて稀少(世界で報告例は数十例規模) |
| 遺伝形式 | 大半が新生突然変異(de novo)。常染色体顕性(優性)形式で稀に遺伝 |
| 主な責任遺伝子 | WDR26、FBXO28、DISP1、HLX、TP53BP2など |
| 国際分類 | ICD-10:Q93.5、Orphanet:ORPHA 250999、GARD:3738 |
1.2 1q43-q44欠失症候群との違い
同じ第1染色体長腕の遠位部にある1q43-q44欠失症候群とは別の疾患です。1q43-q44欠失症候群はAKT3やZBTB18遺伝子の喪失が中心的な役割を果たし、脳梁異常や小頭症を特徴としますが、先天性横隔膜ヘルニアの合併は1q41-q42欠失症候群に特に多いとされています。両者は染色体上で隣接していますが、責任遺伝子も臨床像も異なるため、染色体マイクロアレイ検査による正確な欠失範囲の同定が診断と予後評価において重要です。
1.3 疾患認識の歴史と日本での状況
1q41-q42欠失症候群は、染色体マイクロアレイ検査(aCGH)や全エクソームシーケンス(WES)といった網羅的解析技術の臨床導入に伴って独立した症候群として確立されました。従来のGバンド染色体分染法では微小な欠失を見逃すことが多く、原因不明の発達遅滞として診断されていた症例の中に、本症候群が一定数含まれていたと考えられています。
日本国内でも、Yanagishitaらによる初の確定診断例が報告されており、その特徴的な表現型から臨床的にも認識可能な症候群であることが実証されています。海外では英国のUnique(Rare Chromosome Disorder Support Group)が患者・家族のための情報提供と交流の場を提供しており、医療従事者による臨床ガイドラインの整備も進められています。
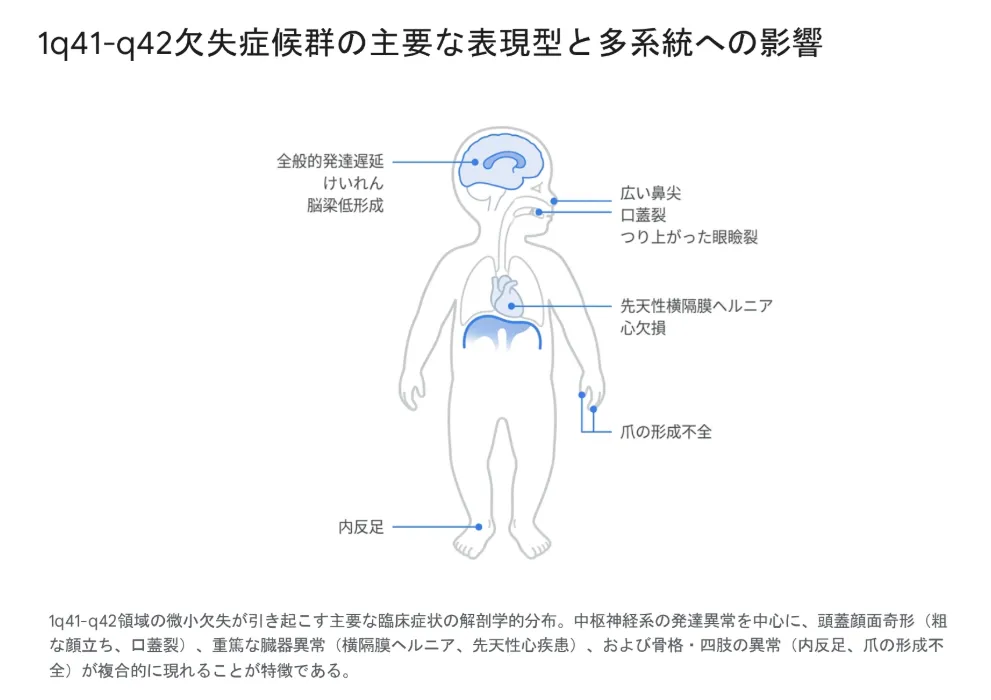
2. 1q41-q42欠失症候群の主な症状|多系統への影響
本症候群は単一の臓器ではなく、中枢神経系・頭蓋顔面・心血管系・骨格系・泌尿生殖器系など多系統に影響します。中でも重度の発達遅滞・知的障害は全例に見られる中核症状であり、先天性横隔膜ヘルニア(CDH)の合併が新生児期の生命予後を大きく左右します。
2.1 主要症状の出現頻度(欠失症候群コホート)
📊 1q41-q42欠失症候群における主要症状の出現頻度
2.2 中枢神経・神経発達への影響
本症候群における神経系への影響は、患者さんの自立度や長期的な生活の質を決定づける中心的な要素です。全例で広範な発達遅滞が認められ、特に言語獲得の遅れや「発語失行」が目立ちます。知的障害の程度は中等度から最重度まで及び、ご家族の生涯にわたる支援が必要となるケースが大半です。
- 発達遅滞:100%の症例で認められる中核症状
- 言語障害:表出性言語の遅れと発語失行が顕著
- てんかん:約63〜95%に発症、点頭てんかんや進行性ミオクローヌスてんかんなど治療抵抗性のケースも
- 脳の構造異常:脳梁の欠損・低形成、全前脳胞症、脳室拡大、Dandy-Walker症候群の合併例など
- 新生児期:顕著な筋緊張低下が哺乳不良の原因に
2.3 特徴的な顔貌(粗な顔立ち)
特異な顔つき(dysmorphic features)は、臨床医が本症候群を疑うための重要な最初のサインとなります。患者さんに共通する顔貌の特徴は、複数の所見が組み合わさって現れます。
- 頭部:前頭部の突出、狭い前頭部、三角頭蓋、長頭症、小頭症
- 眼:深く窪んだ眼、眼裂の斜上、両眼の近接
- 鼻・口:平坦な鼻根、広く前を向いた鼻孔、厚い口唇、短い人中、口蓋裂・粘膜下口蓋裂
- 全体印象:「粗な顔立ち(coarse facial features)」と表現される
2.4 先天性横隔膜ヘルニア(CDH)|新生児期の最重要課題
本症候群において、周産期・新生児期の生命予後を左右する最も重篤な合併症が先天性横隔膜ヘルニアです。患者さんの約18%にCDHが合併すると報告されており、お腹の臓器が胸の中に脱出することで肺が十分に成長できず(肺低形成)、出生直後から極めて重篤な呼吸不全に陥るケースがあります。
・病態:胸とお腹を仕切る横隔膜に穴が開いている状態で、お腹の中の臓器(胃や腸など)が胸に上がってきてしまう先天性の病気です。
・周産期管理:胸の中で肺が十分に成長できず(肺低形成)、出生直後から重い呼吸不全を起こすため、人工呼吸器・一酸化窒素吸入療法、最重症例ではECMOを要します。
・出産計画:胎児期に診断されている場合は、新生児集中治療室(NICU)と小児外科を備えた高次医療機関での出産計画が不可欠です。
2.5 骨格系の特徴|爪の低形成という重要なサイン
本症候群では、著明な低身長、内反足(クラブフット)、第3-4指の合指症、漏斗胸、脊柱側弯症といった骨格の異常が高頻度で見られます。中でも特徴的なのが「爪の低形成・形成不全(nail hypoplasia)」です。
爪の低形成は1q41-q42欠失症候群においては極めて高頻度(100%)に見られる所見であり、臨床診断の重要な手がかりとなります。後述するWDR26単独変異(Skraban-Deardorff症候群)では23%程度と頻度が低いことから、爪の所見は本症候群と単一遺伝子疾患を見分ける重要な鑑別ポイントです。
3. 原因と責任遺伝子|なぜ症状が起こるのか
1q41-q42欠失症候群の症状は、欠失範囲に含まれる複数の遺伝子(WDR26、FBXO28、DISP1、HLX、TP53BP2など)が同時に失われることで生じます。それぞれの遺伝子が異なる役割を担っているため、神経発達障害から内臓奇形まで多臓器に症状が現れます。
通常、私たちの遺伝子は父と母から1コピーずつ、計2コピー受け継いでいます。片方のコピーが欠失または機能しなくなることで、残った1コピーだけでは正常な機能を維持できない状態を「ハプロ不全」と呼びます。本症候群では、欠失領域内の複数の遺伝子が同時にハプロ不全となるため、多臓器に影響が現れます。
3.1 主な責任遺伝子と役割
| 遺伝子 | 主な役割 | 関連症状 |
|---|---|---|
| WDR26 | 神経細胞の発達と機能維持 | 知的障害、てんかん、特徴的顔貌 |
| FBXO28 | タンパク質分解制御、転写制御 | 難治性てんかん、最重度知的障害、多動性運動障害 |
| DISP1 | SHHシグナル経路(胚発生) | 全前脳胞症、CDHの発症リスク |
| HLX | 胚発生・造血系の制御 | 先天性横隔膜ヘルニア、短腸症、無脾症 |
| TP53BP2 | アポトーシス調節(ASPP2) | 脳室拡大、神経管閉鎖障害、心疾患、小眼球症 |
3.2 隣接遺伝子症候群と単一遺伝子疾患の対比|1q41-q42欠失症候群 vs Skraban-Deardorff症候群
WDR26はSkraban-Deardorff症候群の単独責任遺伝子でもあり、1q41-q42欠失症候群と多くの臨床像が重なります。しかし、欠失症候群では複数遺伝子の同時喪失によりCDHや爪低形成などの症状頻度・重症度が大きく異なるため、両者の鑑別は治療戦略と予後予測において極めて重要です。
隣接遺伝子症候群 vs 単一遺伝子疾患|臨床像の比較
3.3 FBXO28|難治性てんかん性脳症の責任遺伝子
FBXO28は近年、非常に重篤な「発達性およびてんかん性脳症(DEE-100)」の単独責任遺伝子として同定された遺伝子です。FBXO28関連てんかん性脳症の臨床像には、最重度の知的障害(WDR26単独変異よりも重症)、生後8ヶ月〜5歳でのてんかん発症(中央値22.5ヶ月)、点頭てんかん・進行性ミオクローヌスてんかんなど薬剤抵抗性の発作型、そして約80%に出現する多動性運動障害といった特徴があります。
興味深いことに、FBXO28遺伝子のコーディング領域(タンパク質配列を決める部分)に変異がない場合でも、同遺伝子の3’非翻訳領域(3′ UTR)の微小欠失が神経発達障害を引き起こした症例も報告されています。これは、遺伝子発現の調節領域も病因となり得ることを示す重要な知見です。
3.4 DISP1とHLX|内臓奇形に関わる遺伝子
DISP1は胚発生で重要な「ソニック・ヘッジホッグ(SHH)シグナル経路」を制御する遺伝子で、全前脳胞症(HPE)や横隔膜ヘルニアの発症リスクに関与します。ただしDISP1単独のハプロ不全だけではこれらの異常を起こすには不十分で、他の遺伝子との相互作用が必要と考えられています。
HLXは「ホメオボックス型転写因子」をコードする遺伝子で、肝臓・胆嚢・腸管・脾臓の発生に必須です。HLXの変異・欠失は先天性横隔膜ヘルニア・短腸症・無脾症を伴う複合先天異常の原因と考えられています。
3.5 遺伝形式と再発リスク
・常染色体顕性(優性):2022年に日本人類遺伝学会で「優性遺伝」が「顕性遺伝」、「劣性遺伝」が「潜性遺伝」へと用語変更されました。本症候群が遺伝するケースでは、片親の片方の染色体に欠失があるだけで子に伝わる可能性がある「常染色体顕性形式」をとります。
・新生突然変異(de novo):両親には欠失がなく、お子さんで新たに突然変異として欠失が発生したケースを意味します。本症候群の大半はこの新生突然変異によって生じます。
本症候群の大半は新生突然変異によって生じるため、次のお子さんへの再発リスクは原則として低いとされています。ただし、ごく稀に、健康な親(軽症で気づかれていなかった保因者)から欠失が遺伝するケースも報告されており、お子さんで欠失が見つかった場合は、両親への検査も検討すべきタイミングといえます。
4. 1q41-q42欠失症候群の診断方法と鑑別診断
確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のGバンド法(核型分析)ではこの微小な欠失を検出することが困難なため、CMAを用いることが現在の診断の標準となっています。出生前と出生後で検査の流れが異なる点も重要なポイントです。
4.1 出生後の確定診断|CMAがゴールドスタンダード
お子さんがすでに生まれており、原因不明の発達遅滞・知的障害・てんかん・先天性奇形などで医療機関を受診した場合、まず臨床評価で本症候群を疑い、血液検体を用いた染色体マイクロアレイ検査を行います。本症候群と確定診断された場合、続いて両親の血液で同じ欠失の有無を確認し(新生突然変異か遺伝かを判定)、頭部MRI、心エコー、腎エコー、眼科・耳鼻科診察、脳波などで合併症の精査を進めます。
CMA(chromosomal microarray analysis)は、従来のGバンド法では検出できない数kb〜数Mb単位の微小な欠失や重複(コピー数変異:CNV)を網羅的に検出する検査です。日本では原因不明の発達遅滞・知的障害・多発奇形に対する保険適用検査として実施されており、1q41-q42欠失症候群の確定診断には欠かせません。
4.2 検査方法ごとの違い
| 検査方法 | 特徴 | 1q41-q42欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 確定診断のゴールドスタンダード。微細CNVを高解像度で検出 | ◎ 確実に検出 |
| Gバンド法(核型分析) | 解像度は約5〜10Mb | ✕ 検出困難(微小欠失は見逃される) |
| FISH法 | 特定領域のプローブで迅速に確認 | △ 専用プローブで可能 |
| 全エクソームシーケンス(WES) | 遺伝子の塩基配列を網羅的に解析 | △ 解析設定によっては可能 |
4.3 鑑別診断|似た症状を示す疾患
1q41-q42欠失症候群は症状が多彩なため、初期評価では他の遺伝性症候群と紛らわしいことがあります。以下のような疾患群との鑑別が重要です。
- Fryns症候群:CDH・肺低形成・粗な顔貌・爪の低形成を四徴とする致死性が高い常染色体潜性(劣性)遺伝疾患。重症の1q41-q42欠失症候群はFryns症候群と臨床的に重なるため、CMAでの鑑別が必須。
- Skraban-Deardorff症候群:WDR26単独変異による疾患。発達遅滞・てんかん・顔貌の特徴は重なるが、爪の低形成や内臓奇形の頻度が異なる。
- DEE-100(FBXO28関連):FBXO28単独変異による発達性およびてんかん性脳症。最重度の知的障害と多動性運動障害が特徴。
- 1q43-q44欠失症候群:同じ1番染色体長腕のより遠位の欠失。AKT3やZBTB18が関与し、脳梁異常・小頭症・知的障害が中心。CDH合併は1q41-q42欠失に特徴的。
お子さんの発達や検査結果が気になっていませんか?
原因不明の発達遅滞や多発奇形には染色体マイクロアレイ検査が有効です。
臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
5. 治療と長期管理|多職種チームでの包括的サポート
1q41-q42欠失症候群には根本的な治療法はまだ存在しません。治療は症状に応じた対症療法・外科的修復・早期療育・継続的支援が中心となり、小児科を司令塔とした多職種チームによる包括的なアプローチが不可欠です。
5.1 急性期|出生直後の救命対応
出生直後に最も迅速な対応が必要なのは、先天性横隔膜ヘルニア(CDH)と重度の先天性心疾患です。CDHが疑われる場合、出生前から新生児集中治療室(NICU)と小児外科を備えた高次医療機関での出産計画が望ましいとされています。
- CDH管理:人工呼吸器、一酸化窒素吸入療法、最重症例ではECMO
- 外科的修復:全身状態安定後にCDHや心疾患の手術
- 栄養管理:哺乳不良に対し経管栄養や胃瘻造設を検討
- 気道管理:Pierre Robin連鎖を伴う場合の気道閉塞対策
5.2 ライフステージ別の管理
| ライフステージ | 主な対応 |
|---|---|
| 新生児期(0〜28日) | CDH・心疾患の救命管理、人工呼吸器、外科的修復、哺乳支援 |
| 乳児期・幼児期(〜5歳) | 早期療育(PT・OT・ST)、口蓋裂手術、てんかんの早期管理、難聴のフォロー |
| 学童期(6〜12歳) | 特別支援教育、骨格異常への装具・手術、てんかん継続管理 |
| 思春期・成人期 | 移行期医療、生活自立支援、就労支援、家族介護負担への支援 |
5.3 てんかんの管理
本症候群ではてんかんの発症率が極めて高く、特にFBXO28関連のケースでは難治性のてんかん性脳症を発症するリスクがあります。標準的な抗てんかん薬による治療で発作コントロールが困難な場合、複数薬の併用、ケトン食療法、迷走神経刺激療法(VNS)などの選択肢があり、てんかん専門医との連携が重要です。小児神経科医による定期的な脳波(EEG)モニタリングが必須となります。
5.4 早期療育とリハビリテーション
重度の発達遅滞・知的障害・運動発達遅滞に対しては、乳幼児期からの早期療育が長期的な発達と生活の質に大きく影響します。可能性を最大限に引き出すため、複数の専門職が連携してサポートします。
- 理学療法(PT):筋緊張低下の改善、内反足のギプス固定後の運動機能獲得、脊柱側弯症の進行予防
- 作業療法(OT):微細運動や食事・着替えなどの日常生活動作(ADL)の習得
- 言語聴覚療法(ST):言語遅滞や発語失行への訓練、代替的コミュニケーション手段(AAC)の導入
- 多職種チーム:臨床遺伝科・小児科・小児外科・小児神経科・小児循環器科・眼科・耳鼻科・心理職・ソーシャルワーカーが連携
5.5 長期予後について
本症候群の長期的な予後は患者さんによって極めて個別的です。新生児期の急性な危機(CDHや致死性心疾患)を乗り越え、致命的な臓器機能不全を伴わないお子さんでは、寿命自体は健常な集団と同等に達する可能性が報告されています。一方で、中等度から重度の知的障害や行動面の課題は永続的であり、生涯にわたる介護や支援を必要とする方が大半です。軽症の親が後に保因者と判明した家族解析では、40代の女性で限定的ながら就労や支援付き住宅での自立生活を送られているケースも報告されており、本症候群の表現型が想像以上に幅広いことを示しています。
6. 遺伝カウンセリングと再発リスク
1q41-q42欠失症候群は表現型の幅が広く、予後予測が容易ではありません。遺伝カウンセリングを通じて、ご家族が病気を正確に理解し、納得のいく決断ができるよう中立的な情報提供を行うことが、医師の重要な役割です。
6.1 カウンセリングで伝えるべきポイント
- 欠失範囲と症状の関係:含まれる遺伝子によって症状の重症度が変わる
- 表現型の多様性:軽症から致死的なものまで幅広いスペクトラムがある
- 予後の不確実性:同じ欠失でも経過は個人ごとに異なる
- 両親の検査:新生突然変異か遺伝かを判定し再発リスクを評価
- 支援体制:多職種チーム、療育、社会福祉制度、家族会の紹介
6.2 再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも欠失なし(新生突然変異) | 原則として低い(1%未満)※生殖細胞モザイクの可能性は残る |
| 片親が保因者 | 理論的に50%(不完全浸透のため、症状の出方は予測困難) |
| 親が均衡型染色体転座 | 転座の種類によりリスクが異なる(個別評価が必要) |
7. 出生前診断とミネルバクリニックのサポート体制
1q41-q42欠失症候群は、NIPTのうち全染色体スクリーニング型のプラン(インペリアルプラン)でリスクを評価でき、羊水検査・絨毛検査でCMAを行うことで確定診断ができます。ただし、出生前に見つけることが常にご家族の利益になるとは限らないため、検査前後の遺伝カウンセリングが不可欠です。
7.1 出生前検査の種類と検出能力
| 検査 | 位置づけ | 1q41-q42欠失への対応 |
|---|---|---|
| NIPT(一般的なターゲット型) | スクリーニング検査 | 対応していないことが多い(特定12微小欠失のみが対象のプランでは対象外) |
| NIPT(全染色体スクリーニング型) | スクリーニング検査 | ○ スクリーニング可能(5Mb以上を対象とするWGS型では1q41-q42領域もカバー) |
| 絨毛検査+CMA | 確定診断 | ◎ 妊娠初期に確定診断可能 |
| 羊水検査+CMA | 確定診断 | ◎ 微小欠失も確定診断 |
7.2 ミネルバクリニックでのNIPTプラン
ミネルバクリニックでは、ご家族のニーズに応じて複数のNIPTプランをご用意しています。ダイヤモンドプランはターゲット法による高精度検査で、特定12箇所の微小欠失(1p36、4p16、5p15、9p、22q11.2など)を高い陽性的中率で検出しますが、1q41-q42はこの12箇所には含まれません。一方インペリアルプランはWGS法とターゲット法のハイブリッドで、5Mb以上の全染色体微小欠失・重複を広範囲にスクリーニングするため、1q41-q42領域もカバーされます。スクリーニング検査のため、陽性時は羊水検査・絨毛検査による確定診断が必要です。
7.3 出生前診断で見つかった場合の対応
出生前に1q41-q42欠失が見つかった場合、本症候群は表現型の幅が非常に広いため、胎児期の超音波所見だけでは将来の予後を正確に予測することが難しい場合があります。遺伝カウンセリングで欠失範囲・関与する遺伝子・表現型の幅・予後の不確実性を中立的に説明し、両親の検査で新生突然変異か遺伝かを判定、詳細超音波で横隔膜ヘルニア・心奇形・脳の構造異常・四肢異常などを精査します。CDHや重度心疾患が疑われる場合はNICUを備えた高次医療機関での出産を検討し、ご家族の不安や葛藤に寄り添い、決断を急がせない時間と環境を確保することが重要です。
⚖️ 倫理的なスタンス|検査は「常に利益」ではない
本症候群のように不完全浸透や表現型の幅が大きい疾患では、出生前に見つけたことが必ずしもご家族の利益になるとは限りません。「特定の検査を勧める」「安心を保証する」「不安をあおる」ような表現は適切ではないと私たちは考えています。検査を受けるかどうか、結果をどう受け止めるかは、十分な情報を得たうえで、ご家族自身が決めるべき事柄です。
7.4 ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。1q41-q42欠失症候群を含む染色体微小欠失症候群について、出生前検査から結果説明、確定検査、その後のフォローまで一貫してサポートいたします。
- 全染色体スクリーニング対応:インペリアルプランでは5Mb以上の全染色体微小欠失・重複を広くスクリーニング、1q41-q42領域もカバー対象
- 確定検査も院内で実施:羊水検査・絨毛検査も院内で実施可能、転院の必要なし
- 臨床遺伝専門医が担当:臨床遺伝専門医が検査前後の遺伝カウンセリングを直接担当
- 互助会で費用面も安心:NIPT受検者全員に適用される互助会(8,000円)により、陽性時の羊水検査費用が全額補助
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
よくある質問(FAQ)
関連記事
参考文献
- OMIM #612530 – Chromosome 1q41-q42 deletion syndrome [外部サイトへ]
- Orphanet – 1q41q42 microdeletion syndrome (ORPHA:250999) [外部サイトへ]
- GARD – 1q41-q42 microdeletion syndrome [外部サイトへ]
- Rosenfeld JA et al. The 1q41q42 microdeletion: variable phenotype and final common pathway. Am J Med Genet A. 2011 [外部サイトへ]
- Yanagishita T et al. 1q41q42 deletion syndrome – First Japanese case report. 2019 [外部サイトへ]
- Skraban CM et al. WDR26 Haploinsufficiency Causes a Recognizable Syndrome (Skraban-Deardorff syndrome). Am J Hum Genet. 2017 [外部サイトへ]
- Schneider AL et al. FBXO28 causes developmental and epileptic encephalopathy (DEE-100). Brain. 2021 [外部サイトへ]
- Slavotinek AM et al. Sequence variants in the HLX gene at chromosome 1q41-1q42 in patients with diaphragmatic hernia. Clin Genet. 2009 [外部サイトへ]
- Chassaing N et al. DISP1 mutations and 1q41-q42 microdeletions in congenital diaphragmatic hernia. Eur J Hum Genet. 2016 [外部サイトへ]
- Vergult S et al. 1q41q42 microdeletion syndrome – phenotypic spectrum. Am J Med Genet A. 2012 [外部サイトへ]
- Au PYB et al. The TP53BP2 gene and 1q41q42 microdeletion syndrome – phenotype expansion. Am J Med Genet A. 2014 [外部サイトへ]
- Cassina M et al. 3’UTR microdeletion of FBXO28 in neurodevelopmental disorder. 2023 [外部サイトへ]
- Unique – Rare Chromosome Disorder Support Group: 1q41-q42 microdeletion factsheet [外部サイトへ]






