疾患概要
Seckel症候群-1(SCKL1)は、染色体3q23に位置するATR遺伝子(601215)のホモ接合体または複合ヘテロ接合体変異によって引き起こされるまれな常染色体劣性遺伝性疾患です。この症候群の特徴には子宮内発育遅延、小人症、精神遅滞を伴う小頭症、そして特徴的な鳥嘴様顔貌が含まれます。Seckel症候群は、遺伝的変異による細胞のDNA修復メカニズムの障害が原因で発生します。これにより、発育に必要な正常な細胞機能が妨げられ、上記のような臨床症状を引き起こすことがあります(Shanske et al., 1997)。
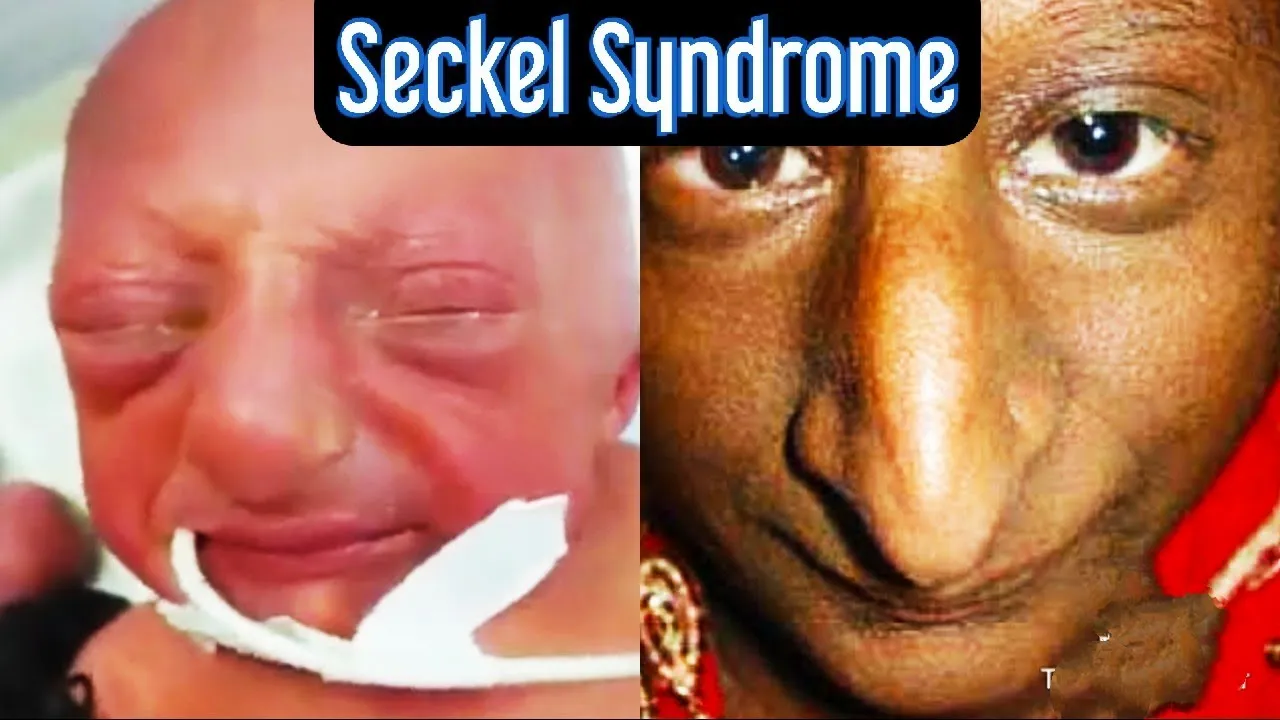
「Bird-headed」鳥嘴様顔貌は、Seckel症候群の特徴的な顔の特徴を指します。この表現は、症候群に伴う顔の特定の外観に由来しており、通常、以下の特徴を含みます。
小さな頭(小頭症): 頭部が通常よりも小さく、比例していない外観を示すことが多いです。
目の大きさ: 目が大きく見えることがあり、顔全体のバランスと比較して不釣り合いに見えることがあります。
鼻の形状: 鼻が尖っていたり、小さかったりすることがあり、これが「鳥のくちばし」に似た外観をもたらすことがあります。
その他の特徴: 顔全体が細長い、または尖った形状をしていることがあります。
これらの特徴は、Seckel症候群の診断を支援する重要な手がかりとなります。ただし、これらの特徴は個人差があるため、症候群の診断には遺伝的検査や他の臨床的評価も重要です。
遺伝的不均一性
SCKL1は、染色体3q23上のATR遺伝子の変異によって引き起こされます。
SCKL2は、染色体18q11上のRBBP8遺伝子の変異によって引き起こされます。
SCKL4は、染色体13q12上のCENPJ遺伝子の変異によって引き起こされます。
SCKL5は、染色体15q21上のCEP152遺伝子の変異によって引き起こされます。これはSCKL5とも呼ばれます。
SCKL6は、染色体3q22上のCEP63遺伝子の変異によって引き起こされます。
SCKL7は、染色体14q22上のNIN遺伝子の変異によって引き起こされます。
SCKL8は、染色体10q21上のDNA2遺伝子の変異によって引き起こされます。
SCKL9は、染色体3p21上のTRAIP遺伝子の変異によって引き起こされます。
SCKL10は、染色体8q24上のNSMCE2遺伝子の変異によって引き起こされます。
また、Kilincらによる2003年の報告では、染色体14q上のセッケル症候群遺伝子座について、SCKL3と命名されましたが、これは後に誤りであることが判明しました。
臨床的特徴
症例について、Seckel(1960)は自身の観察と文献に基づき、決定的な研究を発表しました。また、Harperら(1967)やMajewski and Goecke(1982)、Butlerら(1987)など多くの研究者がセッケル症候群に関する様々な症例を報告しています。特にButlerらは、染色体の不安定性や血液学的な問題を有するセッケル症候群のサブグループの存在を示唆しました。
Hayaniら(1994)は、Seckel症候群と診断された女性のケースを報告し、彼女は26歳で急性骨髄性白血病(AML)と診断されました。これまでにAMLが報告されたことはなかったため、この発見は重要です。
Shanskeら(1997)は、イエメン系アラブ人家族の中で、Seckel症候群を有する3人の兄弟を報告しました。また、Abou-Zahrら(1999)は、Seckel症候群患者の血液学的異常と染色体切断について研究し、ファンコニー貧血との関連を示唆しました。
Goodshipら(2000)は、パキスタンの近親家族で生まれたセッケル症候群の症例を報告しました。この子は、小頭症や歯列叢生、小葉のない小さな耳などの特徴を持っていました。さらに、この症候群はII型小頭症性骨異形成原人小人症(MOPD2)と表現型的に似ていますが、MOPD2はより重度の発育遅延やX線学的異常などによって区別されます。
Canら(2010)は、セッケル症候群の特徴的な特徴を持つ男児を報告し、この子はファロー四徴症などの心奇形を有していました。Ogiら(2012)は、ATR遺伝子の変異によるSeckel症候群-1の症例を報告しました。最後に、Mokrani-Benhelliら(2013)は、出生時に子宮内発育遅延と小人症を示したフランス人女児のケースを記述し、彼女の症状は年齢とともに悪化していました。
多様性
マッピング
分子遺伝学
また、Mokrani-Benhelliら(2013年)の研究では、セッケル症候群を持つ9.5歳のフランス人女児において、ATR遺伝子のミスセンス変異(D1879Y)が見つかりました。さらに、彼女は第3染色体上に540kbの欠失を持ち、この欠失にはATRの他にXRN1、PLS1、TRPC1の3つの遺伝子が含まれていました。この女児の両親は、それぞれ異なる変異をヘテロ接合体(遺伝子の片方が変異している状態)で持っていました。
さらに、セッケル症候群とATRIP遺伝子の変異との関連については、現在も研究が進行中であり、確認待ちの状態です。
ATRIP遺伝子の変異(arg760からterへの置換、R760X)は、セッケル症候群への寄与が明確でないため、意義不明の変異とされています。この変異は、セッケル症候群のグジャラート系インド人の患者においてOgiら(2012年)によって同定されました。この患者は、ATRIP遺伝子のエクソン12においてヘテロ接合性のc.2278C-T転移を持っており、それによってarg760からter(R760X)への置換が生じています。患者の母親もこの変異を持っていましたが、症状はありませんでした。
患者とその両親の細胞に対するRT-PCR解析から、c.2278C-T変異はナンセンス媒介mRNA崩壊(NMD)の対象ではないことがわかりました。父親の対立遺伝子はエクソン2に影響を及ぼすスプライシング欠損を持っており、これはNMDの対象である可能性が高いとされました。この患者には野生型ATRIP活性が約25%残存していると推定されました。ウェスタンブロット分析では、10〜20%の残存タンパク質レベルとATRの減少が示されました。
さらに、この患者の細胞研究とin vitroでの機能発現研究により、R760X変異は紫外線照射後のATR依存性G2/M細胞周期停止を促進せず、ATR-ATRIP相互作用を低下させ、機能喪失と一致することが示されました。臨床的には、この患者は重度の小頭症(-10SD)、小顎症、歯列叢生、小さな耳たぶ、骨年齢の遅延、対称性小人症を有していました。これらの臨床的および細胞的表現型は、ATR変異を有するセッケル症候群患者で観察されるものと類似していました。
動物モデル
この実験で生まれたセッケルマウスは、非常に目立つ小人症の特徴を出生時から示していました。また、これらのマウスの胎盤は壊死(細胞の死)領域が蓄積し、細胞性が全体的に減少していることが観察されました。セッケルマウスは、小頭症や小顎症、後退額などの顔面の異形症も示していました。脳のサイズが小さく、嚢胞があり、脳梁が欠損している特徴もありました。
胚発生期には、セッケルマウスは高いレベルの複製ストレス(DNAが複製する際のストレス)を示していましたが、生後になるとこのストレスは少なくなっていました。それにも関わらず、成体のセッケルマウスは加速した老化の兆候を見せ、特にp53というタンパク質が欠如している場合には、老化がさらに悪化しました。
この研究の結果から、Murgaらは、子宮内での複製ストレスが生後の老化の開始に寄与する可能性があり、これがATRやp53といったチェックポイントタンパク質の複製ストレスを制限する役割とバランスをとることにより、セッケル症候群の症状を説明するモデルを提案しました。
歴史
疾患の別名
SECKEL-TYPE DWARFISM
NANOCEPHALIC DWARFISM
MICROCEPHALIC PRIMORDIAL DWARFISM I
BIRD-HEADED DWARFISM
セッケル型小人症
小頭症
小頭症型小人症I
鳥頭小人症







