目次
先天性角化不全症(DC; dyskeratosis congenita)は、骨髄不全BMF、癌素因、体細胞(非血液)異常を特徴とする遺伝性疾患である。先天性角化症および関連する短テロメア症(telomere biology disorder; TBD)は、テロメア(染色体の末端にあり、核細胞を遺伝物質の損失や増加から保護する領域)の正常な維持を阻害する変異によって引き起こされる。
先天性角化不全症は遺伝性の骨髄不全症候群であり、粘膜白板症、爪の形成不全、皮膚の色素異常の三徴候が典型的な特徴である。罹患者は再生不良性貧血および悪性腫瘍のリスクが高い。一般的でない特徴としては、類上皮腫、早発白髪、小頭症、発達遅延、肺線維症、門脈圧亢進症、免疫不全および胃腸疾患がある。表現型は非常に多様である。すべての罹患者はテロメア維持の欠陥によりテロメアが短縮している(Savageらによる要約、2008年)。
臨床的特徴
先天性角化不全症および関連するテロメア生物学的疾患(DC/TBD)は、テロメア維持の障害により短テロメアまたは極短テロメアを生じる。テロメア生物学的障害の表現型スペクトラムは幅広く、古典的な先天性角化不全症(DC)のほか、テロメアが非常に短く、身体所見が孤立しているものも含まれる。古典的な先天性角化不全症は、爪の形成不全、上胸部および/または頚部のレース状網状色素沈着、口腔白板症の三徴候を特徴とするが、すべての人に認められるわけではない。DC/TBD患者では、進行性骨髄不全(BMF)、骨髄異形成症候群または急性骨髄性白血病、固形癌(通常、頭頸部の扁平上皮癌または肛門性器癌)、肺線維症のリスクが高い。その他の所見として、眼球異常(眼瞼上瞼症、眼瞼炎、睫毛貧毛症、眼瞼外反症、眼瞼内反症、睫毛貧毛症)、歯牙欠損症、肝疾患、消化管毛細血管拡張症、腰や肩の血管壊死などがある。ほとんどのDC/TBD患者は正常な精神運動発達および正常な神経学的機能を有するが、いずれの型においても有意な発達遅滞が認められる。さらに、小脳低形成(Hoyeraal Hreidarsson症候群)および両側滲出性網膜症および頭蓋内石灰沈着(Revesz症候群およびCoats plus症候群)が認められる。DC/TBDの発症および進行は様々であり、軽症の場合は骨髄機能が正常で最小限の身体所見しか認められないものであり、重症の場合は診断上の三徴候および早期発症の骨髄不全BMFを有するものである。
典型的な先天性角化不全症像
当初、DCは臨床的には3つの粘膜皮膚所見によって認識された。
- 皮膚色素異常:上胸部と頚部に網目状の色素沈着。
- 爪ジストロフィー:小さくて薄い爪甲で、縦方向の隆起があり、加齢とともに消失する。手足の爪が侵される。
- 口腔白板症:口腔白板症は口腔粘膜と舌を侵す。
多くの患者では、診断時に3つの特徴すべてが認められるわけではない。患者の約4分の3がこれら3つの古典的症状のうち少なくとも1つを有し、3つの所見のすべてを有する患者は半数よりやや少ない。診断年齢中央値は14歳(出生から75歳までの範囲)である。
骨髄不全
骨髄不全は年齢に関係なく発現し、多くの場合、先天性角化不全症の徴候として現れる。患者の半数近くが40歳までに骨髄不全の徴候を示す。骨髄不全の発症年齢の中央値は10歳であった。
血小板減少と貧血がBMFの最初の徴候であることが多いが、幼児期の細胞減少は必ずしも骨髄形成不全によるものではない。細胞減少症は、免疫調節異常による免疫介在性の場合もあれば、臓器機能不全および/または出血による場合もある。ある症例では、幼少期に単一系統の免疫性細胞減少症と思われる症状を呈し、小児期または青年期に真の汎血球減少症を発症する。
その他の臨床症状
先天性角化不全症および他のテロメア症候群の患者は、様々な臓器症状を含むその他の臨床的特徴を発現する。
いくつかの特徴は典型的に小児期に現れるが、発症年齢も非常に多様である。食道狭窄、涙管破壊、重度の歯・歯周病、免疫不全による再発性感染症、腸症・腸炎、低身長、性腺機能低下、尿道狭窄、網膜症、肺・消化管血管の変化、認知発達障害などの特徴が現れ始めることがあるが、これらの多くは幼児期にはみられない。患者が10歳代以降になると、肺線維症、肝肺症候群や肝硬変などの肝疾患、血管奇形、骨粗鬆症などの骨異常、早発白髪、神経精神障害などのリスクが高まる。
肺線維症と肝硬変は特に注目すべきである。他の多くの臓器異常とは対照的に、一般的に40~50代という成人期に発症し、先天性角化不全症以外では短テロメア症TBDsの初発症状となることがあるためである。肺線維症は先天性角化不全症患者の約5分の1にみられる。特発性肺線維症の家族性症例の15%、散発性症例の5%までが、TERT、TERC、PARN、RTEL1の変異と関連している。
肺線維症と骨髄低形成の組み合わせは、テロメア障害の強力な予測因子として浮上している。骨髄不全と肺線維症の同時発生によりテロメラーゼ変異の存在が増加するかどうかを調べました。これらの基準を満たす連続 10 人、合計 36 人の家族が生殖系列変異テロメラーゼ遺伝子 (100%) を保有していた。
がん素因
古典的先天性角化不全症患者は、多くの種類のがんを発症するリスクが高い。先天性角化不全症におけるがんの発生率は、20歳までは10%未満であるが、50歳までに20~30%に上昇し、最初のがんの診断は中央値で29歳である。
先天性角化不全症と関係するがんとしては以下がある。
頭頸部扁平上皮がん(40パーセント)
胃/食道(約17パーセント)
肛門(12パーセント)
皮膚(12パーセント)
急性白血病(8%;リンパ系より骨髄系が多い)
肝臓(5パーセント)
その他のがんとしては、ホジキンリンパ腫、肺、膵臓、結腸、子宮頸部のがんなどの報告がある。
その他の症候性の疾患
Hoyeraal-Hreidarsson症候群
Hoyeraal-Hreidarsson(ホイエラール・レイダーソン)症候群(HHS)は、小児期早期から発症する臨床的に重症の先天性角化不全症である。
Hoyeraal-Hreidarsson症候群は、ACD、RTEL1、TERT、PARNの劣性遺伝子の変異、およびTINF2の常染色体優性遺伝子の変異により発症する。
先天性角化不全症の古典的な粘膜皮膚および体性特徴に加えて、HHS患者は以下の臨床的特徴を有する。
子宮内発育遅延
小脳低形成
小頭症
発達遅延
重篤な免疫不全(古典的先天性角化不全より悪い)
早期発症進行性骨髄不全
Hoyeraal-Hreidarsson症候群の診断が時代とともに進歩するにつれて、小脳低形成が診断の必要条件となった。
Hoyeraal-Hreidarsson症候群は創始者変異c.3791G>A (p.R1264H)により、アシュケナージ・ユダヤ人を祖先とする個体で高頻度にみられる。
レベス症候群
レベス症候群(RS)は、しばしば重篤なDCの特徴を有することに加え、両側の滲出性網膜症の存在に基づいて定義される。TINF2の変異はRevesz症候群の遺伝的原因として最も多く同定されているが、多くの症例では遺伝的原因はまだ同定されていない。
コートプラス症候群
コートプラス症候群は、滲出液を伴う網膜毛細血管拡張、頭蓋内石灰化、小脳運動障害、骨減少、白質ジストロフィー、成長不良、骨髄異常を特徴とする常染色体劣性遺伝性疾患である。二重鎖テロメアDNAの伸長に関与するCSTテロメア複製複合体の構成要素であるCTC1またはSTN1の常染色体劣性突然変異によって起こる。CTC1変異によるCoats plus症候群の患者ではテロメアがかなり短いが、正常なテロメア長を持つ患者もおり、テロメア短縮とは別のテロメア機能不全がCoats plus症候群の疾患特徴を駆動している可能性が示唆されている。
CTC1変異は古典的なDCの特徴を持つ患者にも見られ、Coats plus症候群と他の短テロメア症候群とのオーバーラップがある。
先天性角化不全症の病態生理
先天性角化症および関連する短テロメア症(TBD)は、テロメアの正常な維持を阻害する変異によって引き起こされる。
テロメアの役割
テロメアは染色体の末端にある特殊な構造体であり、染色体末端の完全性を維持する核酸とタンパク質成分で構成され、染色体の自然末端をDNAの損失や他の染色体との異常な融合から守り、鎖切断によって生じたDNAの遊離末端に反応して通常起こるDNA損傷経路応答の活性化から守っている。テロメアDNAは6塩基のTTAGGG配列のタンデム反復からなる。テロメアDNAのほとんどは二重鎖DNAとして存在し、末端には通常約150から200ヌクレオチドのGリッチ鎖の一本鎖オーバーハングがある。テロメアの二重鎖DNA部分の短縮は、テロメア生物学的障害(TBD)に最も特徴的である。
出生時には、リンパ球などの体細胞のテロメアDNAの長さは8~14キロベース(kb)である。細胞分裂の度に、3’末端の不完全な複製により、テロメアDNAの50から100塩基対が除去される。テロメアが決定的に短い閾値に達すると、細胞はもはや適切な分裂ができなくなり、アポトーシスや老化が起こる。ほとんどの体細胞において、テロメア長の短縮は老化の正常な結果である。一例として、リンパ球のテロメアの平均長は、出生時の約11kbから、100歳ではわずか約4kbまで減少する。このようにテロメアは、細胞分裂の可能な回数を決定する細胞内の「生物時計」と呼ばれている。
ほとんどの体細胞の正常な複製能力が限られているが、常に複製再生を必要とする細胞では、より多くの分裂を可能にするために、テロメアの長さを回復し、より長く維持することができる。例えば、胚性幹細胞、上皮組織や骨髄のようなターンオーバーの激しい組織に存在する多能性幹細胞、悪性腫瘍などではテロメアを合成する能力がある。この複製能力の増大は、テロメラーゼ複合体、つまりテロメアを合成する酵素複合体の作用によって可能になる。テロメラーゼはリボ核タンパク質複合体(RNAとタンパク質)で、染色体の末端にDNAを付加することによってテロメアの短縮に対抗する。テロメラーゼがテロメアを長くする能力は、他にもいくつかのメカニズムによって制御されている。これらのメカニズムに寄与する核タンパク質因子、これらの因子をコードする遺伝子の変異がテロメア制御異常を引き起こす。
- テロメラーゼ
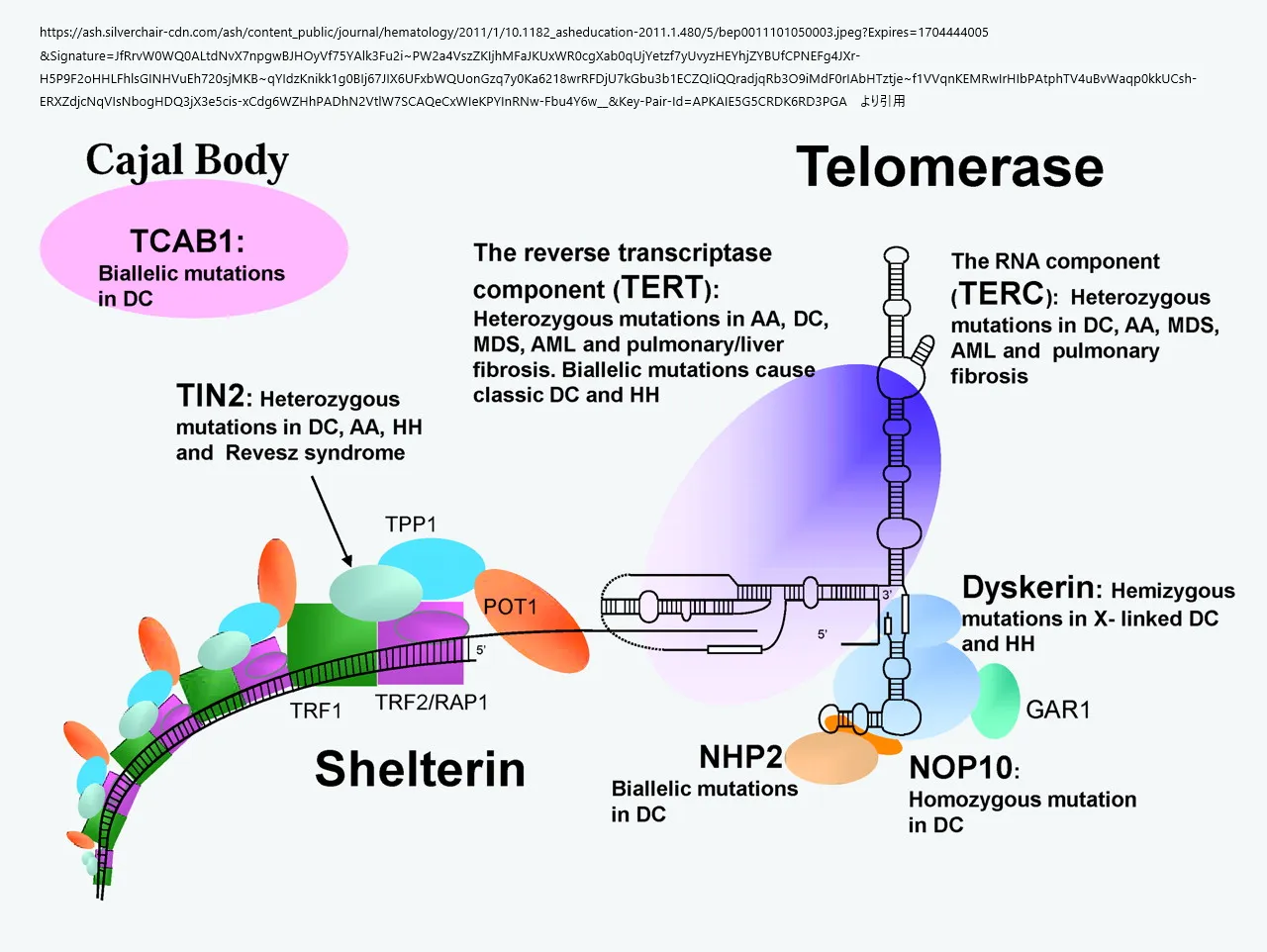
- テロメラーゼは、染色体末端にTTAGGGリピートを付加する逆転写酵素である。触媒タンパク質であるTERTとRNA鋳型であるTRから構成される。テロメラーゼの組み立てと安定性には、dyskerin、NHP2、NOP10を含むいくつかの小核球状リボ核タンパク質(snoRNP)の作用が必要である。NAF1によってコードされるタンパク質は、dyskerinに類似したボックスH/ACA RNA生合成因子であり、TRレベルを制御している可能性がある。
- テロメラーゼの輸送とテロメアへの動員
- 一旦テロメラーゼが組み立てられると、テロメアに動員されなければならない。TRに直接結合するタンパク質TCAB1によって媒介される輸送は、このプロセスにとって重要である。TRF1、TRF2、RAP1、TIN2、TPP1、POT1タンパク質からなるシェルタリン複合体は、t-ループ(TRF2)を作り、安定したテロメアの「キャップ」を作る付加的なタンパク質のリクルートをもたらす機能を果たす。TRF1とTRF2はTNF2をテロメアに動員し、TNF2はTPP1/POT1のヘテロ二量体を動員する。TPP1は次にTELパッチモチーフを介してテロメラーゼをテロメアにリクルートする。
- テロメアの複製
- CTC1/STN1/TEN1(CST)複合体は、テロメラーゼがテロメアDNAのGリッチ鎖を伸長した後、テロメアDNAのCリッチ鎖の伸長を促進し、二重構造のテロメアDNAを形成する。
- テロメアの安定性
- RTEL1 (regulator of telomere length 1)はDNAヘリカーゼであり、二重構造のテロメアDNA複製の完全性と、Dループと呼ばれる構造の解体に寄与する。
TIN2はテロメラーゼのリクルートメントに寄与しているが、TIN2をコードしている遺伝子の変異は、テロメアの維持において、ドミナントネガティブな役割をするがまメカニズムは不明である。さらに、エクソリボヌクレアーゼPARNは他のテロメラーゼ関連因子のRNA転写物の安定性に影響を与える可能性がある。
TBD(telomere biology disorders 、短テロメア症候群またはテロメロパチーとも呼ばれる)では、テロメア機能に関与する因子のいずれかをコードする遺伝子に変異があると、テロメアが異常に短くなる。Dyskeratosis congenita (DC)および関連する短テロメア症候群の原因として知られている変異は、テロメラーゼ活性と輸送、シェルタリン「キャッピング」複合体の形成、テロメアの安定性に関与するタンパク質をコードしている。
罹患していない人のテロメアの正常な短縮速度が年間約60bpであるのに比べ、テロメア障害を持つ人は年間約120bpの割合でテロメアDNAが失われる。さらに、罹患者の次世代は、テロメアが徐々に短くなって生まれる可能性がある短テロメア症候群におけるテロメアの早期短縮は、早期の細胞死、老化、ゲノムの不安定性を引き起こし、その結果、臓器や組織の機能障害、恒常性の変化、不適切な成長につながる。
先天性角化不全症は短テロメア症候群の原型である。先天性角化不全症はいくつかの遺伝性骨髄不全症候群(IBMFS)の一つであり、それぞれの症候群は関連する臨床所見のスペクトルで特徴づけられる。しかし、当時Zinsser-Cole-Engman症候群と呼ばれていた先天性角化不全症の最初の記述は、皮膚所見に焦点を当てたものであった 。その後、皮膚色素沈着異常、爪ジストロフィー、口腔白板症の古典的三徴候が先天性角化不全症を定義するようになった。しかし、よく認識されている他の合併症が、先天性角化不全症の罹患率、特に骨髄不全(BMF)の主な原因であることが多い。
先天性角化不全症や他の短テロメア症候群に加えて、ある種の遺伝性および後天性の癌症候群は、BMFを伴わないテロメラーゼ複合体およびシェルタリン複合体の変異と関連している。例えば、家族性黒色腫、白血病などである。
女性におけるX連鎖性DC/TBDの臨床所見
DKC1病的バリアントのヘテロ接合体である女性における臨床症状は、皮膚色素異常、爪形成異常、骨髄不全などが報告されている。
先天性角化不全症の診断/検査
DC/TBD患者の大多数は、リンパ球サブセットの多色フローサイトメトリー蛍光in situハイブリダイゼーション(flow-FISH)により、年齢の割にテロメアが異常に短いと判定される。現在までに、ACD、CTC1、DKC1、NAF1、NHP2、NOP10、PARN、POT1、RPA1、RTEL1、STN1、TERC、TERT、TINF2、WRAP53、ZCCHC8が、DC/TBDを引き起こし、テロメアが非常に短くなることが知られている遺伝子である。DC/TBDの臨床診断基準を満たす患者の約80%において、これら16遺伝子のいずれかの遺伝子変異が同定されている。
示唆的所見
先天性角化不全症(DC)を含むテロメア生物学的障害(TBD)は、以下の臨床所見を有する個体で疑うべきである。
身体的特徴。以下のいずれか1つ:
- 古典的なDCの臨床的三徴候の少なくとも2つの特徴
- 爪の形成不全。爪の隆起、剥離、成長不良などの軽微なものから、爪のほぼ完全な消失を伴うびまん性のものまである。
上胸部および/または頸部のレース状網状色素沈着。微細またはびまん性の色素沈着亢進または色素沈着低下の可能性がある。異常な色素沈着の変化は上胸部および頸部に限定されないことに注意。
口腔白板症(口腔内の白斑) - 古典的三徴候の1つの特徴または示唆的家族歴(第1度または第2度近親者に骨髄不全[BMF]、骨髄異形成症候群[MDS]、急性骨髄性白血病[AML]、早期発症の頭頸部扁平上皮がん[HNSCC]、および/または肺線維症[PF]の発生)の組み合わせ
- 進行性骨髄不全。年齢に関係なく発現し、また、これが提示徴候となることもある。巨赤芽球症およびヘモグロビンF値の上昇がみられることがある。
MDSまたはAML。臨床症状を呈することがある。
50歳未満で他の危険因子のない人の固形腫瘍、通常は統計部扁平上皮がんまたは肛門性器腺がん。
骨髄不全を発症していない人では、固形腫瘍がDC/TBDの初発症状となることがある。
肺線維症 - 古典的三徴候に加え、以下のうち2つ以上を伴う
- 眼球上視症(眼の過度の充血)。
眼瞼炎(眼瞼上瞼症に起因することが多いまぶたの炎症)
まつ毛の異常成長
早すぎる白髪
脱毛症
歯槽膿漏(歯髄室の拡大)または歯根対歯冠比の低下
発育遅延
低身長
小頭症
性腺機能低下症
食道狭窄
尿道狭窄
肝臓病
骨粗鬆症
腰または肩の血管壊死
消化管毛細血管拡張症
肺動静脈奇形
確定診断
DC/TBDの診断は、特徴的な臨床所見と以下のいずれかを有するプロバンドにおいて確立される。
- 6細胞パネルアッセイにおける自動多色フローサイトメトリー蛍光in situハイブリダイゼーション(フローFISH)によるリンパ球テロメア長検査で決定されるテロメア長の短縮。リンパ球のテロメア長が年齢の第一百分位未満であれば、DC/TBDの感度は97%、特異度は91%である。複合型または非定型DC/TBD患者では、6細胞パネルは、全リンパ球と顆粒球の2パネル検査よりも有益である可能性があるAlter et al 2012。
- 常染色体劣性DC/TBDを単独で引き起こすことが知られている6つの遺伝子のうちの1つにおける両アレルの病原性(または病原性の可能性が高い)バリアントの同定
- 常染色体優性DC/TBDを引き起こすことが知られている5つの遺伝子のうちの1つにおけるヘテロ接合性の病原性(または病原性の可能性が高い)バリアントの同定
- 常染色体劣性および優性DC/TBDに関連する4つの遺伝子のうちの1つにおける一重または二重遺伝性の病原性(または病原性の可能性が高い)バリアント
- X連鎖性DC/TBDの原因として知られているDKC1における半接合性の病原性(または病原性の可能性が高い)バリアント
ACMG/AMPバリアント解釈ガイドラインによると、「病原性バリアント」と「病原性バリアントの可能性が高い」という用語は臨床の場では同義であり、どちらも診断的とみなされ、どちらも臨床的意思決定に使用できることを意味する。
分子遺伝学的検査のアプローチには、表現型に応じて、遺伝子標的検査(多遺伝子パネル)と包括的ゲノム検査(エクソームシークエンシング、ゲノムシークエンシング)を 組み合わせることができる。
遺伝子標的検査では、臨床医がどの遺伝子が関与している可能性が高いかを判断する必要があるが、ゲノム検査ではその必要はない。暗示的所見に記載された特徴的な所見を有する患者は、遺伝子標的検査で診断される可能性が高いが、DC/TBDの診断が考慮されていない患者は、ゲノム検査で診断される可能性が高い。
モザイク
限られた数のDC/TBD患者において、組織限定モザイクが観察されている。末梢血細胞から抽出したDNAの分子遺伝学的検査では観察されなかったTERC生殖細胞系列の病原性変異体が、その人の他の細胞(例えば皮膚線維芽細胞)から抽出したDNAで検出された。同様に、生殖細胞系列のRPA1変異体を持つ個体において、造血系体細胞復帰が報告されている。その結果、これらの細胞から抽出されたDNAから病的バリアントを検出することができなくなった。DC/TBDの診断基準を満たすが、末梢血細胞の分子遺伝学的検査で病原性変異体が同定されない場合には、第二の組織源の分子遺伝学的検査を考慮する必要がある。
先天性角化不全症の遺伝学
テロメラーゼ複合体の構成要素、シェルタリンタンパク質、その他のテロメラーゼ制御因子をコードする遺伝子に変異があることが、DCやその他の短テロメア症候群の個体で報告されている。
先天性角化不全症の診断基準を満たす患者のうち、80%が以下の遺伝子のいずれかに病原性変異を有すると推定されている。
ACD(副腎皮質異形成ホモログ)-常染色体優性遺伝性DC、常染色体劣性遺伝性DC、Hoyeraal-Hreidarsson症候群。ACDはTPP1タンパク質をコードする。
CTC1(保存テロメア維持因子1)-常染色体劣性DC、常染色体劣性Coats plus症候群、石灰化と嚢胞を伴う脳網膜細小血管症(CRMCC)としても知られる。
DKC1-X連鎖性DC、Hoyeraal-Hreidarsson症候群。
NAF1(NEF-associated factor 1; TNFAIP3-interacting protein 1 TNIP1とも呼ばれる) – 常染色体優性遺伝で、肺線維症を含む。
NHP2 (non-histone protein 2; nucleolar protein family A, member 2 NOLA2とも呼ばれる) – 常染色体劣性先天性角化不全症 または肺線維症を含む常染色体優性短テロメア症候群。
NOP10(核小体蛋白質10;核小体蛋白質ファミリーAメンバー3 NOLA3とも呼ばれる)-常染色体劣性先天性角化不全症。
常染色体劣性DC、肺線維症、Hoyeraal-Hreidarsson症候群。
RTEL1(テロメア伸長ヘリカーゼ制御因子1)-常染色体優性遺伝性DC、常染色体劣性遺伝性DC、肺線維症、Hoyeraal-Hreidarsson症候群。
STN1-常染色体劣性Coats plus症候群。
TERC-常染色体優性遺伝DC、肺線維症。
TERT(テロメラーゼ逆転写酵素)-常染色体優性DC、常染色体劣性DC、家族性黒色腫、肺線維症。
TINF2(TRF1-相互作用核因子2)-常染色体優性遺伝DC、Hoyeraal-Hreidarsson症候群、Revesz症候群。
WRAP53(TP53に対するアンチセンスWD反復蛋白;TCAB1蛋白をコード)-常染色体劣性遺伝。
ZCCHC8(テロメラーゼRNAの成熟に必要)-常染色体優性遺伝の肺線維症。
USB1(以前はC16orf57;snRNAの成熟に必要) – バイアレル変異はRothmund Thomson症候群、好中球減少症を伴う多汗症、およびテロメア長は正常であるが先天性角化異常症の表現型を持つ一部の患者と関連している。
MDM4(p53活性亢進)-舌扁平上皮癌や急性骨髄性白血病などの先天性角化不全の特徴を持つ家系において、骨髄不全と短いテロメアに関連する生殖細胞系列のミスセンス変異。
NPM1 – テロメアを調節することなくタンパク質の翻訳を阻害するヘテロ接合性の生殖細胞系列突然変異で発症。
特定の遺伝子の変異は、短テロメア症候群の臨床的影響と分離することが証明されており、個々の遺伝子内の特定の変異の遺伝子型-表現型相関がますます認識されるようになってきている。一般に、先天性角化不全症の臨床的により重篤な変異型は、テロメア長の最大の減少と関連している。
常染色体優性遺伝
ACD、PARN、RTEL1、TERC、TERT、TINF2、NAF1の変異で常染色体優性遺伝が観察されている。
TERCやTERTの変異を持つ患者では、世代を経るにつれて病気の症状が重くなる(表現促進現象)ことがあり、これはテロメアが世代間で徐々に短くなることが原因であると考えられている。TINF2変異を持つ患者はテロメアが極端に短く、5歳以前に骨髄不全を発症することが多い。
常染色体劣性
常染色体劣性疾患は、ACD、CTC1、NHP2、NOP10、PARN、RTEL1、STN1、TERT、WRAP53の変異で観察されている。
X連鎖性
報告されている唯一のX連鎖性DC症候群は、DKC1遺伝子の変異が関与している。ヘテロ接合体の女性では、X染色体の不活性化パターンが歪んでおり、正常なDCK1対立遺伝子を発現している細胞の生存が有利であることが示唆されている 。しかし、6人のヘテロ接合体をレトロスペクティブに解析したところ、5人にDCに特徴的な爪ジストロフィーと皮膚の色素沈着の変化がみられ、2人には臨床的に重大な創傷治癒遅延がみられた。
ashpublications.org/view-large/figure/8182730/bep0011101050003.jpeg
より引用
テロメア維持に関与するテロメラーゼとシェルタリン複合体の模式図。太字のタンパク質/RNA名は、DCおよびその関連疾患において変異している。AAは再生不良性貧血、MDSは骨髄異形成症候群、RSはRevesz症候群を示す。
疫学
先天性角化不全症およびその他のテロメア不全の真の有病率は不明である。明かでない所見を有する患者は、特発性再生不良性貧血(AA)、特発性肺線維症、特発性肝硬変、散発性先天異常などの他の診断を受ける可能性がある。さらに、テロメアの長さに影響を及ぼす遺伝子変異を持つ他の多くの個体は、より微妙な臨床的表現型を持つ可能性がある。極端な例では、死亡率が高いために重篤な先天性角化不全症が過小診断されていることも考えられる。
先天性角化不全症の有病率は一般集団で約100万人に1人と推定されている。骨髄不全患者の約2~5%がDCであることが確認されている。小児は成人よりもDCと診断される可能性が高いが、成人期に肺線維症や肝疾患をきたす他のTBDsが認識されるようになってきている。
管理
症状に対する治療
治療は個々の患者に合わせて行われる。造血細胞移植(HCT)は骨髄不全および白血病に対する唯一の根治的治療法であるが、長期予後は治療毒性により歴史的に不良である。適切なドナーが得られない場合、BMFに対してはアンドロゲン療法が考慮される。その他の癌の治療は、癌の種類に合わせて行われる。特筆すべきは、癌治療は肺や肝の毒性だけでなく、長引く細胞減少のリスクを高める可能性があることである。肺線維症の治療は主に支持療法であるが、肺移植が考慮されることもある。
サーベイランス
- 骨髄不全の場合には、全血球算定(CBC)を正常なら年1回、異常ならそれ以上の頻度で行う。骨髄吸引および生検を年1回行う。アンドロゲン治療を受けている患者では、CBC、肝機能、肝超音波検査、内分泌学的評価の定期的モニタリングを行う。
- がんリスク:口腔がん、頭頸部がんに対する毎月の自己検診、耳鼻咽喉科医と皮膚科医による年1回のがん検診、年1回の婦人科検診。
- 肺線維症:診断時または本人が検査を行えるようになった時(多くの場合~8歳)から年1回の肺機能検査、臨床症状から肺動静脈奇形が疑われる場合は、バブル心エコー検査を行う。
- 6ヵ月ごとの定期的な歯科検診と良好な口腔衛生が推奨される。
- リスクのある親族の評価: 親族にDC/TBDを示唆する徴候や症状がある場合、あるいは造血幹細胞移植のドナー候補として評価されている場合、テロメア長検査、あるいは家系内の病因変異が判明している場合は分子遺伝学的検査が必要である。
遺伝子による表現型の違い
重度の骨髄不全と粘膜三徴候を伴う古典的DCは、常染色体劣性遺伝、X連鎖性遺伝、またはTINF2関連常染色体優性遺伝のDC/TBDと関連している。TINF2を除く常染色体優性遺伝子の変異を有するDC/TBDでは、他の変異を有するDCと比較して、有意に良好な全生存期間が観察される。
常染色体劣性遺伝またはX連鎖性DC/TBDの患者は、常染色体優性遺伝のDC/TBDの患者よりも神経学的所見を示すことが報告されている。
常染色体優性遺伝型のDC/TBDでは、HCTの既往のない肺線維症がより高頻度に報告されている。
遺伝子型-表現型相関
DC/TBDはまれであるため、特定の遺伝子型-表現型相関は同定されていない。
浸透率
DC/TBDおよびDC/TBDに関連する合併症の浸透率はよくわかっていない。同じ家族内でも個人差があり、年齢とともに合併症が増加することから、浸透率は不完全と思われる。最近の研究では、生殖細胞系列の病原性バリアントに対する修飾因子となりうる体細胞TERTプロモーターバリアントが報告されている。

この記事の筆者:仲田洋美(医師)






