疾患概要
www.researchgate.net/figure/Electron-microscopy-results-A-CLN5-disease-juvenile-muscle-biopsy-showing-RL-arrows_fig3_276357766
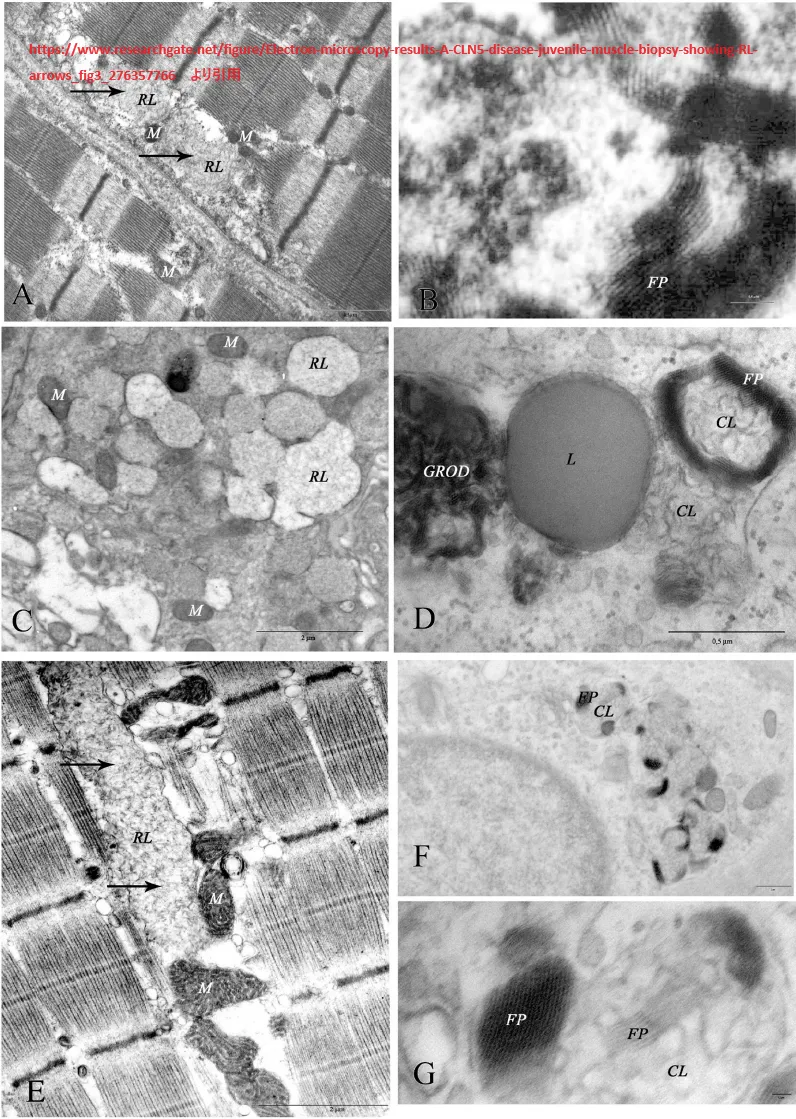
より引用。電子顕微鏡の結果。A.CLN5病(幼若期):筋生検でRL(矢印)を示す(A. L. Taratuto氏提供)。B. 若年性CLN6病:筋生検でFPが密に詰まっている(提供:A. L. Taratuto)。C.若年性CLN7:エクリン分泌腺にRLを伴う小胞を示す皮膚生検。D. CLN8疾患(先天性):皮膚生検で、脂質滴(L)に隣接したGROD、CLおよびFPを示す。E. 若年性CLN3病:筋生検で、CLが主に筋小胞下コンパートメントに認められる(矢印);ミトコンドリアの肥大に注意(M)(提供:A. L. Taratuto)。F.若年性CLN3病:エクリン汗腺にFPとCLが混在する皮膚生検。G.若年性CLN3病(Fの詳細):FPとCLが混在した小体。略号: CLは曲線状小体、RLは直線状小体、FPは指紋状小体、GRODは顆粒状親水性沈着物、Mはミトコンドリオン、Lは脂質滴。
神経細胞性セロイドリポフスチン症(NCL; CLN)は、臨床的および遺伝学的に多様な神経変性疾患の群を形成しており、特定のリポ色素の細胞内蓄積は、これらの疾患を特徴づける重要な要素です。CLNの一般的な表現型と遺伝的不均一性については、CLN1 (256730)を参照してください。
CLN5病は、神経系の進行性疾患であるニーマンピック病タイプC(NCL)の一形態です。NCLは、特定の酵素の欠如または機能不全によって細胞内での材料の蓄積が起こる遺伝子疾患群を指します。CLN5病は、特にCLN5遺伝子の変異によって引き起こされます。
CLN5疾患は遺伝性の神経系に影響を及ぼす病気で、小児期から成人期初期にかけて発症する可能性がありますが、多くの場合、5歳頃に最初の症状が現れます。この疾患を持つ子どもたちは、しばしば正常な発達を遂げているように見えますが、不器用さや以前に獲得した運動技能の喪失(発達退行)といった動きの問題を最初の徴候として経験します。さらに、ミオクロニーてんかん(制御不能な筋肉のピクピクを伴うてんかん発作)、運動失調(動きの協調が困難)、視力低下、言語障害、知的機能の低下などが特徴です。CLN5疾患を持つ患者の平均余命は様々ですが、一般的には青年期や成人期の半ばまで生存することが多いです。
CLN5疾患は、神経細胞セロイドリポフスチン症(NCL)の一種で、バッテン病とも呼ばれる疾患群に属します。NCLは神経系に影響を及ぼし、通常、視覚、運動、思考能力の悪化を引き起こします。NCLは遺伝的原因によってそれぞれ区別され、各サブタイプは「CLN」という名前と、それを示す番号で呼ばれます。これらの疾患は、セロイドリポフスチンと呼ばれる物質の蓄積によって特徴づけられ、特に神経細胞に影響を及ぼします。
CLN5遺伝子には、CLN5病を引き起こす35種類以上の変異があります。この病気は、精神的及び運動機能の発達に障害をもたらし、歩行が困難になったり、知能障害を引き起こしたりします。さらに、患者さんはよくてんかんの発作や視力の低下を経験します。CLN5病の症状は通常5歳ごろに現れますが、青年期や成人期になってから発症することもあります。
この病気を引き起こす変異の多くは、CLN5蛋白質に影響を与え、その前駆体の処理を妨げたり、蛋白質の構造を変えたりします。その結果、蛋白質はリソソームへと運ばれなくなります。他の変異は、異常な蛋白質がすぐに分解されるようになる原因となります。この中の1つの変異は「フィンマジョール」と呼ばれ、フィンランド人におけるCLN5病のほとんどのケースの原因です。フィンマジョール変異は、タンパク質の成分であるチロシンを、蛋白質の生成を早期に停止させるシグナルに置き換えます。
リソソームでCLN5蛋白質が機能しなくなると、特定の蛋白質の分解が妨げられ、細胞全体に蓄積すると考えられます。これらの蓄積が全ての細胞にダメージを与えるものの、神経細胞が特に影響を受けやすいです。CLN5病により神経細胞が大量に失われると、重大な症状が現れ、早死にする原因となります。
青年期や成人期にCLN5病が発症する場合、CLN5遺伝子の変異が原因でCLN5蛋白質の機能が低下し、正常な場合よりも速く分解されると考えられています。変異したCLN5蛋白質が短時間しか機能しないため、損傷した蛋白質や不要な蛋白質がリソソームで分解されることがあります。これらの物質が蓄積し、神経細胞の死を引き起こすまでには時間がかかるため、このような患者の症状は人生の後半に現れます。
遺伝的不均一性
臨床的特徴
フィンランドの研究
Santavuoriらによる研究は、フィンランド西海岸の限定された地域で後期小児神経性セロイドリポフスチン症(一般にバッテン病として知られるNCLの一形態)の集団を特定しました。この研究は、疾患の臨床的特徴を明確にし、特に精神発達障害、運動失調、ミオクロニーてんかんなどの特徴を報告しました。また、この疾患が古典的な乳児後期障害と類似しているが、発症年齢がやや遅く、生存期間が長いことを発見しました。
コロンビアの研究
Pineda-Trujilloらによる報告は、コロンビアの血族家族内で若年型変型NCLを発症した2人の兄姉に焦点を当てました。この研究は、視力障害、筋力低下、下肢の振戦など、病状の急速な進行を示しました。行動変化、言語喪失、ミオクローヌス、てんかん発作も観察されました。皮膚生検の電子顕微鏡検査により、変型NCLを示唆する指紋状封入体が見つかりました。
多様な民族的背景を持つ患者の研究
Xinらの研究は、非フィンランド系白人、フランス系カナダ人、ヒスパニック、スウェーデン人、中国人、アジア系インド人、エジプト人、パキスタン人など、多様な民族的背景を持つCLN5患者10例を報告しました。これは、CLN5病が特定の民族や地域に限定されないことを示しています。発症年齢は4歳から17歳で、臨床的特徴には大きなばらつきがありましたが、多くは運動障害、てんかん発作、視覚障害を経験しました。
これらの報告から、NCL、特にCLN5病は高度に異質な疾患であり、発症年齢、臨床的進行、生存期間に大きな変動があることがわかります。電子顕微鏡検査により、封入体の形状にも多様性があることが示されています。これらの発見は、CLN5病の診断と治療における課題を強調しており、さらなる研究が必要であることを示しています。
臨床的バリエーション
Manciniら(2015)の研究は、CLN5疾患における臨床的変異の範囲を広げる興味深い例です。通常、CLN5疾患は幼少期から青年期にかけて発症するとされていますが、この研究は50代半ばで発症した2人の兄弟のケースを報告しています。これらの患者は小脳失調と進行性の認知障害を主な症状として示し、不安定歩行、眼振、振戦、三半規管運動失調、測定障害、運動障害、反射亢進などの神経学的徴候が見られました。特に一人の患者では、脳画像において小脳と皮質の萎縮が著明であることが示されています。これらの症状と画像所見は、CLN5疾患が引き起こす神経細胞の損失と脳の構造変化を反映しています。
全ゲノム配列決定により、両兄弟はCLN5遺伝子においてホモ接合性のミスセンス変異(S312N; 608102.0009)を持っていることが明らかになりました。Manciniらはこの変異を「hypomorphic allele」と推定しています。Hypomorphic alleleは、野生型アレルと比較して、タンパク質の活性が低下したり、タンパク質の量が少なくなったりする変異を指します。このような変異は、タンパク質の機能が完全には失われていないため、疾患の発症が遅れたり、症状の程度が軽減されたりすることがあります。このケースでは、CLN5タンパク質の活性の部分的な保持が、発症の遅れと比較的緩やかな進行性を説明している可能性があります。
この報告は、CLN5疾患の臨床的なスペクトラムが想定されていたものよりも広いことを示唆しています。また、遺伝的変異の特性が疾患の表現型にどのように影響するかを理解する上で重要な情報を提供しており、特定の変異が疾患の進行に与える影響を予測する手がかりを与えるかもしれません。さらに、このような例は、遺伝子療法や他の分子的介入戦略の開発において、標的とする遺伝子変異の特性を考慮する必要性を強調しています。
命名法
この命名法は、疾患の理解を深め、様々な遺伝的変異によって引き起こされる病態の特定を容易にします。それにより、患者の診断、治療法の選択、および将来の研究の方向性に役立てることができます。CLN5を含む神経細胞性セロイドリポフスチン症の分類には、遺伝的原因の特定が重要であり、それが治療や研究における新たな可能性を開く鍵となります。
マッピング
Williamsら (1994) は、11家族における13人の罹患児と17人の健常兄弟を対象にした連鎖研究を行い、CLN5遺伝子座を1p上のCLN1と16p上のCLN3(204200)の両方から除外できることを発見しました。
Savukoskiら (1994) は連鎖分析を用いてCLN5遺伝子座を13q21.1-q32にマッピングしました。この研究では、疾患対立遺伝子とD13S162およびD13S160のマーカーとの間に高いレベルの連鎖不平衡があることを示しました。
さらに、Klockarsら (1996) はこの位置をさらに絞り込み、マーカーCOLAC1とAC224の間の約350kbの領域にマッピングしました。
これらの研究は、CLN5遺伝子の正確な位置を特定し、フィンランド型後期幼児型NCLの遺伝的基盤の理解を深める上で重要な役割を果たしました。これにより、遺伝子検査や治療法の開発に向けた研究が進む基盤が築かれました。
遺伝
両親は、それぞれ変異した遺伝子の1つのコピーを持っていますが、もう1つのコピーが正常であるため、通常はこの疾患の徴候や症状を示しません。これらの個体は「キャリア」と呼ばれ、疾患を発症することはありませんが、変異遺伝子を次世代に伝える可能性があります。
両親がそれぞれ変異した遺伝子のキャリアである場合、それぞれの妊娠において以下の確率が生じます。
子供が変異遺伝子のコピーを2つ受け継ぐ(一つは母親から、もう一つは父親から)確率は25%で、この場合、子供は疾患を発症します。
子供が変異遺伝子の1つのコピーのみを受け継ぎ(つまり、キャリアになる)確率は50%です。この場合、子供は疾患を発症しませんが、将来自分の子供に変異遺伝子を伝える可能性があります。
子供が変異遺伝子のコピーを受け継がない確率は25%で、この場合、子供は疾患のキャリアでもなく、疾患を発症することもありません。
常染色体劣性遺伝疾患の診断と管理は、遺伝カウンセリングを含むことが多く、家族が遺伝的リスクを理解し、将来の妊娠における遺伝的選択肢を検討できるようにします。
頻度
CLN5病の発症率はまだ完全には明らかにされていませんが、科学文献には85例以上の症例が記載されています。当初、この病気はフィンランド人に特有のものと考えられていました。これは、フィンランドではNCL疾患群、特にCLN5病が比較的高い頻度で見られるためです。フィンランドでは約12,500人に1人がNCLを患っていると推定されています。
しかし、時間が経つにつれて、CLN5病がフィンランド人だけでなく、世界中の多様な人々に影響を与える可能性があることが明らかになりました。これは、CLN5病が特定の人種や民族に限定されるものではなく、広範な地理的分布を持つことを示しています。
全てのNCLを合わせた場合、世界中で推定10万人に1人が何らかの形のNCLを患っているとされています。これにはCLN1からCLN14までのさまざまなタイプが含まれ、それぞれが異なる遺伝子の変異によって引き起こされます。
NCLの形態によっては、幼児期から成人期にかけて発症することがあり、進行性の視覚障害、認知障害、運動機能の低下、てんかんなど、さまざまな症状が現れます。現在、これらの疾患の根本的な治療法は存在せず、治療は症状の管理に焦点を当てています。
CLN5病のような希少疾患の研究は、新しい治療法の開発に不可欠です。遺伝子療法や細胞治療などの新しい治療アプローチが、将来的にはこれらの疾患を治療する可能性があります。
原因
CLN5遺伝子の変異は、このタンパク質の構造を変え、リソソームへの到達を妨げることが多いです。結果として、リソソーム内で正常に機能するCLN5タンパク質が不足し、特定のタンパク質の分解が妨げられ、細胞内に蓄積します。この蓄積は細胞にダメージを与え、特に神経細胞が傷つきやすいことから、神経系の広範な損失につながり、重篤な徴候や症状を引き起こし、早期の死に至ることがあります。
青年期や成人期に発症するCLN5疾患の場合、変異したCLN5タンパク質が正常なものよりも早く分解されると考えられています。このため、損傷したタンパク質や不要なタンパク質がある程度はリソソームで分解される可能性がありますが、これらの物質の蓄積が神経細胞死を引き起こすまでには時間がかかります。その結果、このタイプのCLN5疾患を持つ患者では、徴候や症状が人生の後半に現れることがあります。
CLN5疾患の治療には現在、症状を管理し、患者の生活の質を改善するための対症療法が中心です。この疾患に特異的な治療法はまだ確立されていませんが、遺伝子療法や小分子薬剤の開発など、研究が進められています。
分子遺伝学
●主な発見
Savukoskiら(1998): CLN5遺伝子に3つの異なる変異を同定。これらはCLN5病の原因となる最初の遺伝子変異の一部であり、特にフィンランド人患者において同定されました。
Pineda-Trujilloら(2005): CLN5遺伝子のホモ接合体変異をコロンビアの血族家族で同定。この発見は、疾患が北欧以外の地域にも存在することを示しました。
Xinら(2010): フィンランド人以外の47人の患者から14の変異を同定し、その中には11の新規変異が含まれていました。これらの変異は、タンパク質の早期終止を引き起こすことが多いと報告されました。
El Haddadら(2012): CLN5遺伝子にホモ接合性の切断型変異を同定し、CLN5病がCLN9疾患として誤分類されていた患者を再分類しました。また、CLN5-null細胞はセラミド合成酵素の活動に影響を及ぼし、これが疾患の病態生理に関連している可能性が示唆されました。
●分子遺伝学の意義
これらの研究は、CLN5病の遺伝的多様性と複雑さを示しています。特に、CLN5遺伝子の異なる変異が世界中のさまざまな人口に存在することは、疾患の診断と理解に重要な意味を持ちます。また、これらの研究は、疾患の病態生理の理解を深め、将来的にはより効果的な治療法の開発につながる可能性があります。
特に、セラミド合成酵素の活動とCLN5病の関連は、疾患の治療に新たなアプローチを提供するかもしれません。セラミド合成経路の調節に焦点を当てた治療戦略が、将来的にはCLN5病患者に有益である可能性があります。これらの分子遺伝学的研究は、希少疾患の理解と治療の進歩に不可欠です。
集団遺伝学
この研究により同定されたハプロタイプは、D13S160とD13S162というフランキングマーカーによって定義され、有病染色体の大部分(81%)に存在していました。さらに、マーカーD13S162の対立遺伝子4は、有病染色体の94%で観察されました。これらの結果は、CLN5疾患に関連する遺伝子変異がこの地域の特定の祖先から派生したことを示唆しています。
連鎖不平衡の解析は、CLN5変異が比較的新しい出来事であることを支持しました。連鎖不平衡は、遺伝子座間の物理的距離が近い場合に、遺伝子座が一緒に継承される傾向を指し、特定の遺伝子変異の周りに特定のハプロタイプパターンが保存される現象です。この研究では、11cMの遺伝的距離にわたる7つのマーカーで連鎖不平衡が検出され、これはCLN5変異が約500年前、すなわち20~30世代前に発生したことを示唆しています。
系譜研究と連鎖不平衡の結果を組み合わせることで、VariloらはCLN5遺伝子座がマーカーD13S162から約200〜400kbの距離に位置すると予測しました。この精密な位置情報は、CLN5疾患の原因となる遺伝子の同定と理解に向けた重要なステップを提供しました。フィンランドのCLN5患者集団におけるこの創始者効果の発見は、遺伝性疾患の研究において、地理的、歴史的文脈がどのように遺伝的変異の分布に影響を与えるかを理解する上での一例となっています。
動物モデル
電子顕微鏡による蓄積物質の観察では、フィンガープリントプロファイル、曲線体、直線体など、ラメラ構造のプロファイルが混在していることが明らかになりました。さらに、特定の脳領域においてGABA作動性介在ニューロンのサブセットの顕著な消失が報告されました。これは、特定の神経細胞群がこの疾患によって特に影響を受けることを示しています。
このモデルマウスにおける脳内転写産物のプロファイリングは、神経変性、防御・免疫反応に関与する複数の遺伝子の発現変化を明らかにしました。また、ミエリンの構造成分の発現低下が検出され、これはヒトのCLN5患者におけるミエリン形成不全と一致しています。
顕著な脳の萎縮を示さなかったことから、KopraらはこのCln5 -/- マウスモデルが、老化に関連する分子プロセスの研究に役立つ可能性を示唆しました。このモデルはCLN5病の理解を深めるだけでなく、病態メカニズムの解明や新たな治療法の開発に貢献する重要なツールとなります。
疾患の別名
●代替タイトル;記号
CEROID LIPOFUSCINOSIS, NEURONAL, 5, VARIABLE AGE AT ONSET
セロイドリポフスチン沈着症, 神経細胞性, 5歳 発症年齢不詳
●この項目に含まれる他の疾患
NEURONAL CEROID LIPOFUSCINOSIS, LATE INFANTILE, FINNISH VARIANT, INCLUDED
FINNISH vLINCL, INCLUDED
神経性セロイドリポフスチン症、乳児期後期、フィンランド変種、含む
フィンランド変異型、含む







