疾患概要
ceroid lipofuscinosis-3 (CLN3)
Ceroid lipofuscinosis, neuronal, 3 新生児セロイドリポフスチン症3 204200 AR 3
神経細胞性セロイドリポフスチン症-3(CLN3)、通称バッテン病、は染色体16p12に位置するCLN3遺伝子(607042)の変異によって引き起こされる遺伝性疾患です。この病気は、ホモ接合体または複合ヘテロ接合体の変異により生じ、主に小児期に神経系の退行を引き起こします。
神経細胞性セロイドリポフスチン症(NCL; CLN)は、進行性の神経変性を引き起こす一群の遺伝性疾患です。これらの疾患は、細胞内に自家蛍光性リポ色素蓄積物質が異なるパターンで蓄積することによって特徴づけられます。NCLは臨床的にも遺伝学的にも多様であり、進行性の痴呆、てんかん発作、進行性の視覚障害などの症状を含む臨床経過をたどります。
特に、CLN3型のNCLは、「フィンガープリント」プロファイルと呼ばれる特有のリポ色素の超微細構造パターンを示します。このプロファイルは、リソソーム残渣体内に純粋に存在するもの、曲線状または直線状のプロファイルを伴うもの、および大きな膜結合リソソーム液胞内に小さな成分として存在するものの3つの異なる形態を呈することがあります。CLN3患者の血液リンパ球内で見られるフィンガープリントプロファイルの組み合わせは、この病態の典型的な特徴です。
NCLの遺伝的不均一性は広範にわたり、異なる遺伝子変異が異なる型のNCLを引き起こします。たとえば、CLN1型は特定の遺伝子変異に関連していることが知られています。これらの疾患は、特に小児期に発症し、進行性の神経機能障害を引き起こすことが多いため、診断と管理が特に重要です。NCLの研究は、これらの複雑な疾患の理解を深め、将来的には治療法の開発につながる可能性があります。
CLN3遺伝子の変異は65種類以上確認されており、これらが原因で神経細胞性セロイドリポフスチン症-3(CLN3病)を引き起こします。この遺伝性疾患は小児期に発症し、主に神経系に影響を与え、視力障害、知的障害、運動障害、言語障害、てんかん発作の悪化などの症状を引き起こします。特に多くの症例で見られる変異は、CLN3遺伝子から約1,000塩基対が欠失する1kb欠失と呼ばれるもので、これにより異常に短いタンパク質が産生され、細胞プロセスに影響を及ぼします。このタンパク質の欠損や機能不全がCLN3病の症状を引き起こす正確なメカニズムは未解明です。
CLN3疾患の患者では、リソソーム内に蛋白質やその他の物質が蓄積し、これが全身の細胞、特に神経細胞で起こります。これらの蓄積は細胞損傷や細胞死を引き起こし、神経細胞の死が進行すると神経学的症状が現れます。しかし、CLN3遺伝子の変異がリソソームの異常蓄積にどのように関与しているのかは、まだ明らかにされていません。この病態の解明は、将来的な治療法の開発に向けた重要なステップとなります。
CLN3疾患は、進行性の遺伝性疾患であり、主に神経系に影響を及ぼします。この病態は、神経細胞性セロイドリポフスチン症(NCL)の一種であり、一般にバッテン病としても知られています。CLN3疾患は、特定の年齢での発症と、症状の進行性の悪化によって特徴づけられます。
CLN3疾患の主な特徴と症状の概要:
初期の発症: 通常、4~6歳の間に正常な発育の後、視力障害が初めて現れます。これは、網膜変性によるもので、目の奥の光に敏感な組織が破壊されます。視力障害は徐々に悪化し、多くの患者は小児期後半から青年期にかけて失明します。
認知機能と学習の障害: 子供が4~8歳の頃には、学習遅延が始まり、新しい情報を学ぶことが困難になり、以前に習得した能力の喪失が見られます。発達退行が進行し、通常は完全な文章での会話能力が失われます。
てんかん発作と運動障害: 思春期になると、てんかん発作や運動異常が現れることが多く、筋肉の硬直やこわばり、不器用さ、動作の緩慢さや低下などが含まれます。罹患者は時間と共に歩行や座位の自立が困難になり、車椅子を必要とするようになります。
精神病と幻覚: 現実の歪んだ見方や誤った認識を起こすことがあり、これは精神病や幻覚の形で現れることがあります。
不整脈: 一部の患者では、心臓のリズムに異常が生じることがあります。
CLN3疾患は、特定の遺伝的原因によって引き起こされるNCLのサブタイプの一つです。各NCLサブタイプは、遺伝子の異なる変異によって区別され、セロイドリポフスチンの蓄積という共通の病態生理を持ちながらも、発症年齢や進行速度、症状の種類において異なります。CLN3疾患の患者は、一連の挑戦的な症状に直面し、治療は症状の管理と生活の質の向上に焦点を当てた支持療法が中心となります。
遺伝的不均一性
臨床的特徴
Batten (1903, 1914):
家族性の若年発症「黄斑変化を伴う大脳変性症」を報告。
Spaltonら(1980):
26人のバッテン病患者を検討し、6〜7歳で急速に進行する視覚障害、早期の精神低下、てんかん発作、黄斑変性が初期の一貫した特徴であることを確認。
Wisniewskiら(1992):
若年発症CLN患者163人の臨床的および病理学的特徴を検討し、視力低下、てんかん発作、進行する精神・運動機能障害が特徴であることを報告。
Boustany(1992):
57例のCLN3患者をレビューし、視力低下に続いて認知機能の低下が進行し、ほとんどの患者が10歳までにてんかん発作を発症することを報告。
Taschnerら(1995)、International Batten Disease Consortium (1995):
CLN3遺伝子の変異に焦点を当て、視力低下、てんかん発作、運動障害などの臨床的特徴を報告。
Wisniewskiら(1998)、Corteseら(2014)、Nielsenら(2015):
CLN3の遷延型を含む、若年性および成人発症型NCL患者の臨床的特徴について報告し、視力低下、てんかん発作、認知および運動機能障害、白内障および緑内障の発症などの特徴を確認。
これらの研究は、バッテン病が複雑な疾患群であること、そして患者が経験する一連の挑戦が病態の進行に伴って変化する可能性があることを示しています。バッテン病の臨床的管理には、患者とその家族に対する総合的なサポートと、可能であれば症状の軽減を目指した介入が必要です。
病理学的特徴
バッテン病(ニューロナルセロイドリポフスチン症、NCL)の病理学的特徴は、神経細胞の広範な変性と死、特に網膜と脳実質における重度の喪失、およびリポフスチンの蓄積によって特徴づけられます。リポフスチンは、リソソームに蓄積する脂質とタンパク質の混合物であり、細胞老化のマーカーとして知られていますが、バッテン病においては特に異常な蓄積を示します。
病理学的特徴の詳細については、以下のように要約できます:
神経細胞の広範な変性と死:バッテン病の進行とともに、特に脳実質と網膜において、神経細胞が広範囲にわたって変性し、死に至ります。これは、機能障害や視覚障害など、バッテン病の臨床的症状に直接寄与します。
リポフスチンの蓄積:バッテン病患者の神経細胞周囲にリポフスチンが蓄積します。この物質の蓄積は、神経細胞の機能障害や死に寄与し、病態の進行を促進します。
リンパ球の空胞化:ホモ接合体患者ではリンパ球の空胞化が確立された特徴であり、これは一部のヘテロ接合体保因者でも観察されますが、その頻度や程度は異なります。
白血球ペルオキシダーゼの欠損:CLN2およびCLN3の患者において白血球ペルオキシダーゼの欠損が報告されていますが、この所見は一貫して確認されているわけではありません。
甲状腺および皮膚の異常:バッテン病患者の甲状腺にリポフスチンの蓄積が見られることがあり、皮膚生検では特有の超微細構造が観察されます。
ミトコンドリア異常:特にCLN2およびCLN3患者では、ミトコンドリア内膜のATP合成酵素複合体の特定のサブユニットの異常な蓄積がリソソームで認められます。
アルギニン輸送の欠損:若年性バッテン病のリンパ芽細胞株では、リソソームがアルギニンの輸送に欠損を示し、これが疾患の特定の病態に関与している可能性が示唆されています。
これらの病理学的特徴は、バッテン病の診断、理解、および治療戦略の開発において重要な役割を果たします。特に、リポフスチンの蓄積や特定の細胞内変化の検出は、疾患のバイオマーカーとして利用され得ます。
命名法
神経細胞性セロイドリポフスチン症(NCL)の命名法は、疾患の理解が進むにつれて進化してきました。当初、NCLは発症年齢に基づいて分類され、CLN3疾患は若年発症型(Juvenile Neuronal Ceroid Lipofuscinosis, JNCL)と呼ばれ、4歳から10歳の間に発症することが特徴でした。しかし、遺伝子変異の同定により、NCLの分類法は変化し、現在では基礎となる遺伝子の異常に従って数値で分類されています。したがって、CLN3疾患は、発症年齢にかかわらず、CLN3遺伝子の変異によって引き起こされるNCLを指します。
CLN3疾患は、バッテン病、Vogt-Spielmeyer病、Spielmeyer-Sjogren病としても知られています。これらの名称は、疾患の特定の臨床的特徴や歴史的な発見を反映していますが、「バッテン病」はNCLの総称として広く用いられるようになりました。Moleらによる1999年の定義によれば、NCLは多くの細胞型に自家蛍光性リポ色素の蓄積を特徴とする神経変性疾患群とされています。
この進化した命名法と分類法は、NCLの診断、研究、および治療において、遺伝的特性を基にしたより精密なアプローチを可能にしています。遺伝子変異に基づく分類は、患者の臨床的管理とカスタマイズされた治療戦略の開発に役立っています。
マッピング
研究の進展について要約すると:
Eibergら (1989): ハプトグロビン(HP)とバッテン病の間に染色体16q22上で連鎖が見出されました(最大lodスコア3.00)。
Gardinerら (1990): バッテン病の16番染色体へのマッピングを確認しました(マーカーD16S148においてlodスコアの最大値は6.05)。
Callenら (1991): CLN3の最も可能性の高い位置はD16S67とD16S148の間であると結論づけました。この区間は16p12.3-p12.1と16p12.1-p11.2に物理的にマッピングされました。
Mitchisonら (1993): 70のCLN3家系における研究で、CLN3はD16S297とD16S57の間に局在することがわかりました。ハプロタイプ解析から、CLN3染色体の大部分は単一の創始突然変異から生じたと示唆されました。
Lernerら (1994): ジヌクレオチド反復マーカーを用いて、CLN3の位置を16p12.1に絞り込みました。D16S298/D16S299ハプロタイプがCLN3染色体の54%を占め、これは対照染色体の8%と比較して非常に過剰に存在していました。
Mitchisonら (1995): フィンランドの27家系を含む111の罹患家系の研究で、CLN3遺伝子はD16S298から8.8kb、D16S299から165.4kbの範囲に存在すると予測されました。特定の対立遺伝子の濃縮から、バッテン病の原因はフィンランドでも他のほとんどのヨーロッパ諸国と同様に、同じ主要な突然変異であることが示されました。
これらの研究は、遺伝子マッピングの努力を通じて、CLN3遺伝子の正確な染色体位置を特定し、バッテン病の分子生物学的基盤を明らかにするための基礎を築きました。これらの発見は、遺伝子治療や他の治療戦略の開発に向けた重要なステップです。
遺伝
CLN3型バッテン病に関するこの知見は、診断、遺伝カウンセリング、および将来の治療戦略の開発において重要です。遺伝カウンセリングでは、バッテン病を持つ子供を持つリスクがある家族に対して、遺伝的リスク、疾患の伝達方法、および家族計画に関する情報が提供されます。
また、CLN3型バッテン病の病因として特定された遺伝子変異の特定は、この病気の分子生物学的基盤の理解を深めるのに役立ちます。これにより、病気の進行を遅らせるか治療するための標的となる分子や経路の特定につながる可能性があります。さらに、遺伝子治療や他の分子治療戦略の開発においても、この遺伝的情報は極めて価値があります。
総じて、International Batten Disease ConsortiumによるCLN3の伝播パターンの報告は、バッテン病の遺伝学、診断、および治療において重要なステップを表しています。
頻度
NCLの有病率は地域によって異なり、全体としては比較的まれな疾患です。一般的に、すべてのNCLのタイプを合わせた有病率は、世界中で約10万人に1人と推定されています。しかし、特定の地域や集団では、遺伝的な要因によって有病率が高くなることがあります。例えば、特定のNCLの形態はフィンランドなどの国でより一般的であり、”フィンランド型”として知られることもあります。
CLN3疾患の正確な有病率に関するデータは限られており、研究によって異なることがあります。病気のまれな性質と診断の困難さが、有病率の正確な推定を複雑にしています。遺伝的スクリーニングと診断技術の進歩により、NCLとそのサブタイプの理解が深まり、より正確な有病率の推定が可能になることが期待されます。
NCLの治療法は現在のところ根治的なものではありませんが、症状の管理と患者の生活の質の向上を目指した支援が行われています。また、遺伝子療法や細胞治療など、将来的な治療法の開発に向けた研究が進行中です。
原因
CLN3遺伝子の変異は、タンパク質の構造や量に影響を及ぼし、結果的に異常なタンパク質が産生されます。これにより、タンパク質が適切に機能せず、細胞内のプロセスが阻害される可能性があります。特に、異常なCLN3タンパク質の存在は、リソソームの機能不全を引き起こし、リソソーム内にタンパク質やその他の物質が蓄積することにつながります。
この蓄積は、特に神経細胞において、細胞損傷や細胞死を引き起こす可能性があります。神経細胞の死が進行すると、CLN3疾患の典型的な神経学的徴候や症状が現れます。これには、視覚障害、認知障害、運動機能の低下、そしててんかん発作などが含まれます。
CLN3遺伝子の変異がリソソームへの物質の蓄積にどのように関与しているのか、またその蓄積がどのようにしてCLN3疾患の症状を引き起こすのかについては、まだ完全には解明されていません。研究者たちは、この疾患の発症メカニズムを解明し、将来的に治療法を開発するために、CLN3タンパク質の機能やリソソームの機能不全についての理解を深めるための研究を続けています。
診断
イミダゾールアミノ酸尿症の同定: BessmanとBaldwin (1962) は、非血縁家系の患者とその近親者にイミダゾールアミノ酸尿症が見られることを報告しました。この発見は、ヘテロ接合体の検出や疾患の異質性の同定に役立つ可能性があると示唆されました。
メタクロマシアに基づく同定: DanesとBearn (1968) は、ホモ接合体とヘテロ接合体を、細胞培養した皮膚線維芽細胞のメタクロマシアに基づいて同定できることを発見しました。
低分子量ペプチドの検出: LaBadieとPullarkat (1990) は、ポリアクリルアミドゲル電気泳動により、バッテン病患者の尿中に低分子量ペプチドが存在することを証明しました。これは疾患の特異的生化学的マーカーとなる可能性があります。
超微細構造検査: Goebel (1996) は、病型の正確な診断には患者の細胞の超微細構造検査が必要であると指摘しました。特にCLN3疾患は、指紋プロファイルによって特徴づけられます。
遺伝子検査の開発: Jarvelaら (1996) とTaschnerら (1997) は、CLN3遺伝子の特定の変異や欠失を検出するための迅速な遺伝子検査法を開発しました。
統一バッテン病評価尺度 (UBDRS): Marshallら (2005) は、若年発症CLN3患者の運動能力、行動能力、機能的能力を評価するための臨床評価尺度を開発しました。
これらの進歩により、NCLの診断はより正確で迅速に行えるようになり、患者の管理と治療戦略の策定に役立っています。特に遺伝子検査の開発は、疾患の早期診断と家族歴の評価に重要な役割を果たしています。
- フィンガープリントプロファイル
-
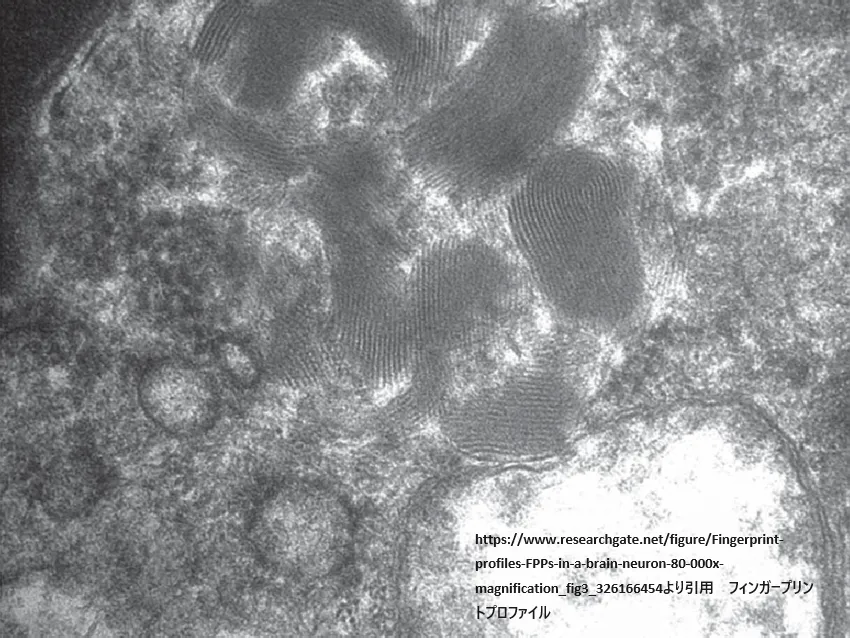
https://www.researchgate.net/figure/Fingerprint-profiles-FPPs-in-a-brain-neuron-80-000x-magnification_fig3_326166454
より引用
CLN3疾患における「フィンガープリントプロファイル」とは、細胞内に蓄積する特有のリポ色素蓄積物質の超微細構造パターンを指します。このパターンは電子顕微鏡で観察され、特徴的な指紋のような模様が見られることからこの名前がつけられました。フィンガープリントプロファイルは、神経細胞性セロイドリポフスチン症(NCL)の中でも特にCLN3型(若年発症型)に関連して観察されます。リポ色素蓄積物質は、細胞のリソソーム内に異常蓄積する自家蛍光性物質であり、セロイドリポフスチンとしても知られています。これらの蓄積物は、神経細胞を含む体の多くの細胞タイプで見られることがあります。CLN3疾患におけるこれらの蓄積物の超微細構造は、病態の理解や診断の際に重要な手がかりを提供します。
フィンガープリントプロファイルの存在は、CLN3疾患の診断を支援し、他の型のNCLや他の神経変性疾患との鑑別に役立ちます。この特徴的なパターンを持つリポ色素蓄積物質の同定は、患者の細胞の超微細構造検査を通じて行われ、CLN3疾患の特定において重要な役割を果たします。
治療・臨床管理
レボドパは、パーキンソン病の治療に一般的に使用される薬剤で、ドパミン前駆体として作用します。体内でドパミンに変換され、ドパミン不足に関連する症状を緩和します。この研究では、レボドパを受けた10人の患者のうち6人が1年後に症状の減少を示し、4人は症状の増加を示しましたが、そのうち3人は6ヵ月後に症状の減少を示しました。これは、レボドパが若年発症CLN3の患者における錐体外路症状の管理において一定の効果を示す可能性があることを示唆しています。
セレギリンは、モノアミンオキシダーゼB(MAO-B)阻害剤であり、脳内のドパミンの分解を遅らせることでドパミンの作用を延長させます。この研究では、セレギリン治療を受けた患者と未治療の患者の間に1年後に有意差は認められなかったことから、セレギリンがこの特定の状況で有効な治療オプションであるかどうかは不明確です。
この研究から得られる知見は、若年発症CLN3の臨床的管理において、ドパミン作用薬が一部の患者に対して有益な効果を提供する可能性があることを示しています。しかし、セレギリンについては、追加的な研究が必要とされる可能性があります。これらの結果は、バッテン病の複雑な臨床像を管理するための治療選択肢を拡大するための一歩となり、患者の生活の質を向上させるための新たな可能性を開くものです。
分子遺伝学
1.02-kb欠失の発見: International Batten Disease Consortiumによる1995年の研究では、バッテン病染色体の73%を占める主要な変異がCLN3遺伝子の1.02-kb欠失であることが明らかにされました。フィンランドでは、この特定の欠失がバッテン病患者の90%に見られるという報告があり、非常に高い割合であることが示されました。
地理的変異: さまざまな地理的背景を持つ患者群で異なる変異が同定されています。例えば、モロッコのバッテン病患者では、Taschnerらによって異なる小さな欠失のホモ接合性が発見されました。これは、CLN3遺伝子の変異が世界中で多様であることを示しています。
変異の同定: Munroeらによる1997年の研究では、188人のバッテン病患者のうち74%が1.02kbのCLN3欠失のホモ接合体であることが明らかにされ、19の新規変異が同定されました。これにより、CLN3における既知の疾患関連変異の総数が23に達しました。
変異の集計: Moleらは1999年に、CLN3に関連する25の変異と2つの多型を報告しました。これらの集計作業は、特定の変異が特定の地理的または人口集団においてより一般的であるかどうかを理解するのに役立ちます。
これらの研究は、バッテン病の診断、遺伝カウンセリング、および将来の治療法の開発において重要な役割を果たします。遺伝子変異の同定は、患者とその家族に対する正確な診断と情報提供を可能にし、また、遺伝子療法や他の分子的介入戦略のターゲットとなる可能性のある特定の分子経路の理解を深めます。バッテン病のような遺伝性疾患に対する治療法の開発には、このような分子遺伝学的知識が不可欠です。
遺伝子型と表現型の関係
この研究によると、1.02kb欠失はCLN3タンパク質の残存機能を保持しており、この変異型タンパク質を過剰発現させるとリソソームのサイズが一貫して減少します。これは、変異型CLN3タンパク質が依然として重要な細胞機能を果たしていることを示唆しています。さらに、この変異型タンパク質の大部分が小胞体内に保持されることから、その機能の一部が細胞内の特定の局所に限定されている可能性があります。
マウス細胞モデルと酵母を使用した実験では、対応する変異転写産物が重要な機能を保持していることが確認されました。これは、特定の遺伝子変異がCLN3疾患の臨床的な症状にどのように影響を与えるかを理解する上で重要な意味を持ちます。
Kitzmullerらの研究結果は、JNCLが変異特異的な疾患表現型を持つこと、そしてCLN3変異の中でも特定の変異が残存機能を通じて疾患の進行や重症度に影響を与える可能性があることを示しています。このことは、JNCLの患者において疾患の発症が遅れることや、臨床症状が比較的軽度であることの説明になり得ます。
この研究は、CLN3疾患の分子生物学的な理解を深めるとともに、将来の治療戦略の開発に向けて重要な洞察を提供しています。変異特異的な治療アプローチが可能であることを示唆し、特定の遺伝子変異をターゲットとした治療法の開発につながる可能性があります。
集団遺伝学
一方、CLN3はフィンランドにおいて特に多く見られることが知られています。Mitchisonらによる1995年の研究では、フィンランドにおけるCLN3の発生率が出生21,000人に1人、保因者頻度が70人に1人であると推定されています。このように、CLN3疾患の発生率と保因者頻度は、地理的な位置や遺伝的背景によって大きく異なる可能性があります。
これらの集団遺伝学的なデータは、特定の遺伝性疾患がどの程度一般的であるか、また特定の集団におけるリスクがどの程度高いかを理解するのに重要です。また、これらの情報は、疾患のスクリーニングや予防策、さらには遺伝カウンセリングの戦略を立てる際にも役立ちます。特にフィンランドのように疾患の発生率が高い地域では、集団遺伝学的な情報が公衆衛生上の対策や研究の指針となり得ます。
動物モデル
ヒツジとイヌのモデル: Jollyら(1992)によるヒツジのバッテン病モデルと、Koppang(1992)およびTaylor and Farrow(1992)によるイヌのモデルは、疾患の自然発生型を研究する上で貴重なリソースです。これらのモデルは、バッテン病の臨床的特徴や進行を理解する上で役立ち、疾患のメカニズムや治療戦略の開発において重要な情報を提供します。
酵母モデル: PearceとSherman(1998)によって開発された酵母モデルは、CLN3遺伝子の研究において特に重要です。彼らはCLN3遺伝子のSaccharomyces cerevisiaeホモログであるBTN1をクローニングし、酵母とヒトのタンパク質間の相同性を利用してバッテン病の基本的な生物学的過程を解明しました。このモデルは、ヒトの疾患と直接的な関係を持つ分子メカニズムの研究を可能にし、特に、CLN3タンパク質の機能や疾患における役割の理解を深めました。
マウスモデル: Chattopadhyayら(2002)によるCln3ノックアウトマウスモデルは、バッテン病の免疫学的側面を探る上で新たな視点を提供しました。彼らの研究は、GAD2に対する自己抗体の存在がバッテン病における神経変性の特徴であることを示し、GABA作動性ニューロンの喪失に寄与する可能性のある自己免疫反応を明らかにしました。この発見は、バッテン病の治療法の開発に向けた新たなアプローチを示唆しています。
これらの動物モデルを通じて、バッテン病の複雑な病理学的過程が明らかにされ、将来的な治療法の開発に向けた基盤が築かれています。特に、異なるモデル間での相互作用や相補性を理解することは、疾患の包括的な理解に不可欠です。
歴史
この転座は、患者の母親や姉妹には認められず、父親は解析不能であったため、この染色体の異常がバッテン病の直接的な原因であるかどうかは明確ではありませんでした。Tuck-Mullerらは、この転座がバッテン病とは無関係である可能性があると述べましたが、この所見はバッテン病の遺伝的不均一性の可能性を示唆しています。
バッテン病、および広義のNCLは、多様な遺伝的変異によって引き起こされることが知られています。これらの遺伝子変異は、病気の特定のサブタイプを定義し、それぞれが異なる臨床的表現を示します。Tuck-Mullerらによる研究は、NCLに関連する遺伝的背景の複雑さを浮き彫りにし、特定の染色体異常が疾患の発症にどのように関与するか、または関与しないかについてのさらなる調査の必要性を示しています。
このような遺伝的研究は、NCLのより良い理解と将来の治療法の開発に向けた基礎を築くものです。遺伝的不均一性の認識は、疾患の診断、管理、および患者とその家族へのカウンセリングにおいて重要な意味を持ちます。
疾患の別名
BATTEN DISEASE
VOGT-SPIELMEYER DISEASE
SPIELMEYER-SJOGREN DISEASE







