疾患に関係する遺伝子/染色体領域
疾患概要
Adrenal hyperplasia, congenital, due to 21-hydroxylase deficiency 21-ヒドロキシラーゼ欠損症による先天性副腎過形成 201910 AR 3
Hyperandrogenism, nonclassic type, due to 21-hydroxylase deficiency 21-ヒドロキシラーゼ欠損症による非典型的な高アンドロゲン血症 201910 AR 3
先天性副腎過形成(CAH)は、副腎でコルチゾールを生産する過程における酵素の欠如によって引き起こされる一群の遺伝性疾患です。この疾患の大部分(約95%)は、21-ヒドロキシラーゼと呼ばれる酵素の機能不全に関連しています。この酵素の活動は、染色体6p21に位置するCYP21A2遺伝子によってコードされ、この遺伝子のホモ接合体または複合ヘテロ接合体変異がCAHの原因となります。
21-ヒドロキシラーゼの欠損により、17-ヒドロキシプロゲステロン(17-OHP)が11-デオキシコルチゾールに変換されず、コルチゾールの合成が妨げられます。これにより、副腎皮質刺激ホルモン(ACTH)のレベルが上昇し、コルチゾールの前駆体である17-OHPが過剰に産生され、蓄積します。これによって副腎からのアンドロゲンの過剰産生が促され、男性化(女性や男性の早期または過剰な第二次性徴)などの症状が引き起こされます。
Slominskiら(1996)の研究では、副腎だけでなく皮膚でもCYP21A2、CYP11A1(コレステロール側鎖切断酵素をコード)、CYP17(17α-ヒドロキシラーゼ/17,20リアーゼをコード)、ACTHR(副腎皮質刺激ホルモン受容体をコード)遺伝子が発現していることが明らかにされました。これらの発見は、これらの遺伝子が皮膚の生理学および病理学において重要な役割を果たしている可能性を示唆しています。また、皮膚でプロオピオメラノコルチン(POMC)が活性化されることにより、局所的に合成されるグルココルチコイドを介したフィードバック機構が存在する可能性が示されています。これは、皮膚が単なる外部のバリアではなく、ホルモン合成と調節においても活動的な役割を果たしていることを示しています。
21-ヒドロキシラーゼ欠損症は、CYP21A2遺伝子に起こる100以上の異なる変異によって引き起こされる遺伝性疾患です。この遺伝子は21-ヒドロキシラーゼ酵素をコードしており、この酵素は副腎でコルチゾールとアルドステロンの生産に必要です。変異の一部は、6番染色体上のCYP21A2遺伝子とその近くにある偽遺伝子(機能しないDNA断片)との間で遺伝物質が交換される「遺伝子変換」というプロセスによって生じます。偽遺伝子からCYP21A2遺伝子へエラーが導入されると、正常なタンパク質の生成が妨げられ、21-ヒドロキシラーゼの欠損が引き起こされます。
21-ヒドロキシラーゼ欠損症には、以下の3つの主な型があります。
1. 塩類消耗型: この型では、CYP21A2遺伝子の変異が酵素の完全な機能喪失を引き起こし、最も重篤な形態です。塩分のバランスの維持が困難となり、重篤な電解質の不均衡や脱水症状を引き起こすことがあります。このタイプは最も重篤で、ホルモン産生の極端な低下を引き起こします。罹患者はナトリウムを多量に尿中に失い、乳児期早期には生命を脅かす症状が現れることがあります。
2. 単純男性化型: CYP21A2遺伝子に変異があり、限定的ですがある程度機能する酵素が生成されます。この型では、塩類消耗型ほど重篤ではありませんが、成長と発達に影響を及ぼす可能性があります。この形態は軽症で、塩分喪失は起こりませんが、男性化の症状が見られます。
3. 非古典型: 最も軽症であり、時には症状が全く現れないこともあります。変異により酵素の産生量は減少しますが、他のどの型よりも多くの機能的酵素が残ります。症状はより軽度で、しばしば成人期まで診断されないことがあります。非典型型の女性は女性型の生殖器を持ち、年齢とともに多毛症、男性型脱毛症、月経不順、生殖能力の低下などの症状が現れることがあります。非典型型の男性では、早期のひげ成長や小さな精巣が見られることがあります。
これらすべての型で、コルチゾールとアルドステロンの産生が阻害され、これらのホルモンの前駆体が副腎に蓄積され、代わりにアンドロゲンへと変換されます。アンドロゲンの過剰な産生は、外性器の男性化や早熟など、性的発達の異常につながります。
21-ヒドロキシラーゼ欠損症の管理と治療は、症状の重症度と患者の個々のニーズに基づいていますが、通常、ホルモン補充療法を含みます。適切な治療により、多くの患者は正常またはほぼ正常な生活を送ることができます。
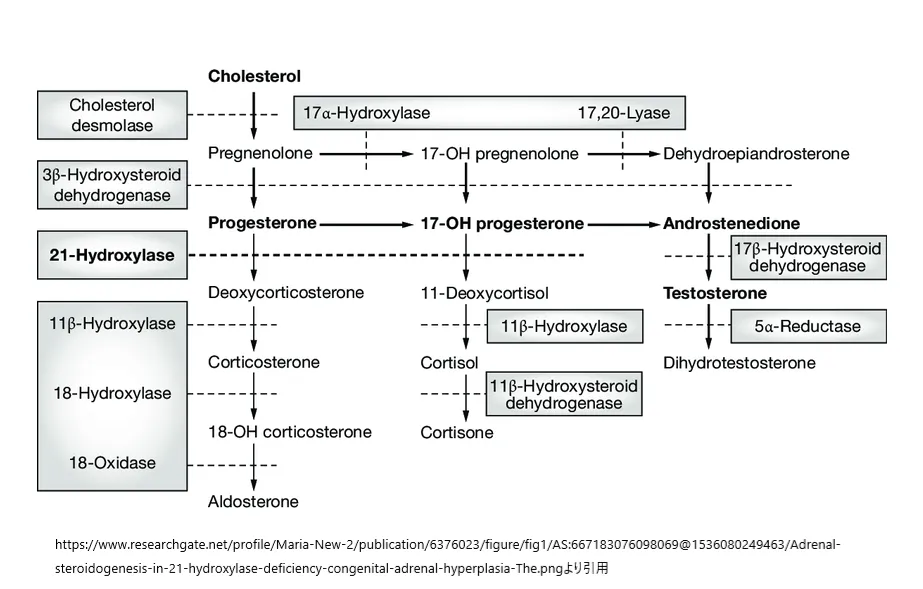
https://www.researchgate.net/figure/Adrenal-steroidogenesis-in-21-hydroxylase-deficiency-congenital-adrenal-hyperplasia-The_fig1_6376023
より引用
遺伝的不均一性
臨床的特徴
新生児期の女性では、外性器の男性化が見られる一方で、生殖腺と内性器は正常です。未治療の場合、男性および女性は急速な成長、性器の増大、早期の骨端閉鎖、そして低身長を経験することがあります。21-ヒドロキシラーゼ欠損症の軽症型は成人になってから発症することがあり、最も軽症の場合は多毛症のみが現れます。
21-ヒドロキシラーゼ欠損症の2つの臨床型(食塩喪失型と非食塩喪失型)は、コルチゾール経路の欠損の程度と相関しているとされています。一部の研究では、21-水酸化系に2つの活性部位が存在するとされていますが、別の研究ではこれを否定し、塩喪失型では両方の部位が欠損し、非塩喪失型では1つの部位のみが欠損すると示唆しています。
21-ヒドロキシラーゼ欠損症に関連する特異な症状として、女性化乳房や両側精巣腫瘍が報告されています。知能に関しては、副腎性器症候群の患者が平均より高いIQを持つという報告がありましたが、これに対して異なる結果を示す研究も存在します。全体として、先天性副腎過形成は多様な臨床像を持つ疾患であり、患者によって発症形態や症状の重さが異なります。
Blanksteinら(1980年)は、21-ヒドロキシラーゼ欠損症の対立遺伝の可能性がある28歳と30歳の2人の姉妹を報告しました。これらの姉妹は原発性不妊症と軽度の多毛症を示していましたが、思春期の発達は正常であり、月経も規則的で女性性徴も正常でした。彼女たちのきょうだいは健康で、罹患した兄弟姉妹はHLA型が同一であるのに対し、健康な兄弟姉妹は異なるHLA型を持っていました。
Levineら(1980年)は、古典的な21-ヒドロキシラーゼ欠損症患者124家族の血清アンドロゲンおよび17-ヒドロキシプロゲステロンレベル、およびHLA遺伝子型を調査し、「隠微性21-ヒドロキシラーゼ欠損症」という状態を報告しました。これらの患者は過剰な男性化、無月経、または不妊症の臨床症状を伴わずに21-ヒドロキシラーゼ欠損症の生化学的証拠を持っていました。これらの患者が古典的遺伝子と隠微遺伝子の複合ヘテロ接合体であることが示唆されました。
Duck(1981年)は、ミルウォーキーで治療を受けた21-ヒドロキシラーゼ欠損症の小児患者42人(36家族)の中で、4人が悪性腫瘍(肉腫または星細胞腫)を発症したことを報告しました。
Kuttennら(1985年)は、400人の女性のうち24人(6%)で21-ヒドロキシラーゼ欠損症が多毛症の基礎であることを発見しました。診断は、血漿中の17-ヒドロキシプロゲステロン濃度の上昇と、ACTH刺激後の顕著な増加に基づいていました。この状態の臨床的発現は、アンドロゲンに対する皮膚の感受性によって決定される可能性があることが示唆されています。これらの発見は、21-ヒドロキシラーゼ欠損症が特定の臨床的症状を示すが、その発現には遺伝的および生物学的な多様性があることを示しています。
Knochenhauerら(1997年)の研究は、CYP21変異のヘテロ接合性が女性における臨床的に明らかなアンドロゲン過剰症のリスクを増加させるかどうかを調べましたが、その結果、ヘテロ接合体がアンドロゲン亢進症のリスクを明確に増加させるとは結論付けられませんでした。ただし、CYP21の欠損は平均テストステロン値および遊離テストステロン値の上昇と関連していることが見られました。
Sinnottら(1989年)は、HLA多型と21-ヒドロキシラーゼおよびC4遺伝子座の遺伝子構成を分析し、同じHLAを持つ21-ヒドロキシラーゼ欠損症の兄弟姉妹間で臨床的特徴に深い不一致があることを発見しました。これは、21-ヒドロキシラーゼ欠損症の臨床的表現には、遺伝子だけでなく他の要因も関与している可能性があることを示唆しています。
Jareschら(1992年)は、21-ヒドロキシラーゼ欠損症の患者において、ホモ接合体およびヘテロ接合体の状態に関わらず、無症候性副腎腫瘍の頻度が高いことを発見しました。これは、副腎腫瘍が偶然に発見された場合でも、常に先天性副腎過形成(CAH)を除外する必要があることを示唆しています。
Ravichandranら(1996年)は、ホモ接合体およびヘテロ接合体の先天性副腎過形成患者では副腎の断面積が増大し、副腎偶発腫の有病率が増大することを指摘しました。特に、単純男性化型CAHや性分化異常を持つ女性で副腎偶発腫が見られた場合、悪性化の可能性は低いものの、CAHの診断を含めた詳細な評価が推奨されます。
これらの研究は、21-ヒドロキシラーゼ欠損症が持つ複雑な遺伝学と臨床的表現の多様性を浮き彫りにし、遺伝的背景だけでなく、臨床的アプローチや評価の重要性を強調しています。
Beuschleinら(1998年)の研究では、21-ヒドロキシラーゼ欠損症が副腎皮質腫瘍の発生に関与している可能性が示唆されました。彼らはアルドステロン産生腺腫、コルチゾール産生腺腫、非機能性偶発腺腫、副腎癌のサンプルを分析し、CYP21B遺伝子の変異スペクトルとP450c21のmRNA発現を調査しました。発見された変異には、val281からleuへのヘテロ接合性変異やエクソン3の体細胞性微小欠失が含まれていました。この研究は、21-水酸化酵素欠損が副腎腫瘍形成の主要因ではない可能性を示唆していますが、腫瘍の臨床的表現型とP450c21遺伝子の発現との間に相関があることを発見しました。
Stikkelbroeckら(2001年)は、思春期および成人男性CAH患者における精巣腫瘍の有病率を調査し、超音波検査によってほとんどの患者で精巣腫瘍が検出されたことを報告しました。治療不足が見られた患者では腫瘍がより大きく、精巣副腎皮質腫瘍がしばしば精子形成障害とライディッヒ細胞不全を伴うことが分かりました。
Urbanら(1978年)は、先天性男性化副腎過形成症の男性における21-水酸化酵素欠損症の有病率を追跡調査し、ほとんどの患者にこの欠損症が存在することを発見しました。
Ghizzoniら(1997年)は、非クラシックCAHの思春期前の小児と健常な低身長の小児を対象に、コルチゾールとTSHの分泌パターンを評価しました。彼らはTSHとコルチゾールの分泌に負の相関があり、軽度の副腎機能障害でさえTSH分泌に影響を与えうることを発見しました。
Meyer-Bahlburg(1999)は、古典的先天性副腎過形成(CAH)の女性において受胎率が比較的低いことを指摘しました。Mulaikalら(1987)の研究では、古典的21水酸化酵素欠損症(CAH)を持つ80人の女性(SV型とSW型に均等に分かれる)について調査し、その半数が異性交遊の経験がないにもかかわらず、異性交遊が活発な女性でも生殖能力が低い傾向にあることが示されました。十分な膣再建手術と異性交遊の両方を報告したSV型の女性では受胎率が60%であるのに対し、SW型の女性ではわずか7%であったことが示されました。
Merkeら(2000)は、先天性副腎過形成患者において、血漿中のエピネフリンおよびメタネフリン濃度と尿中のエピネフリン排泄量が、副腎クリーゼで入院経験のある患者ではそうでない患者に比べて約50%低いことを発見しました。両側副腎摘出術を受けた患者では、副腎髄質の形成不全とクロマフィン細胞の分泌小胞の枯渇が観察され、先天性副腎過形成が副腎髄質系の発達と機能の両方に影響を及ぼすことが結論付けられました。
Green-Golanら(2007)は、古典的CAHの青年と対照群との比較研究を行い、中強度運動時の血糖コントロールの欠陥、代謝およびホルモン反応の変化をCAH患者で確認しました。
Nordenstromら(2002)は、CAHの女児が男性的なおもちゃで遊ぶ傾向があることを発見し、この行動の男性化が疾患の重症度と用量反応関係を示すことを明らかにしました。これは、出生前のアンドロゲン暴露が人間の脳に組織的影響を及ぼし、性徴行動のある側面を決定することを示唆しています。
Merkeら(2003)は、古典的CAHの小児を対象に脳のMRI研究を行い、扁桃体積の有意な減少を確認しましたが、CAH患者の海馬には異常が見られませんでした。これは、出生前のグルココルチコイド欠損症が扁桃体の成長と発達に特に影響を及ぼすことを示しています。これらの研究結果は、CAHに関連する身体的および行動的特徴、および患者の生理学的および神経発達的影響に関する重要な洞察を提供します。
BerenbaumとBailey(2003年)の研究では、先天性副腎過形成(CAH)を持つ女児の性自認について調査し、その結果、性自認が性器の男性化の程度や性器再建手術を受けた年齢とは関連していないことが明らかにされました。彼らは、発育初期の中等度のアンドロゲン過剰が非典型的な性同一性のリスクをわずかに増加させる可能性があるものの、このリスクは性器の男性化からは予測できないと結論付けています。この研究は、性自認が単純な生物学的要因だけでなく、より複雑な心理的、社会的要因によっても形成されることを示唆しています。
Gidlofら(2007年)は、重度のCYP21欠損症を持つ女性患者が軽症の患者よりも妊娠期間が長いことを発見しました。これは、アンドロゲン過剰、17-ヒドロキシプロゲステロン濃度の上昇、コルチゾール欠乏、またはこれらの因子の組み合わせが妊娠期間の延長に重要な役割を果たす可能性を示しています。この研究は、ステロイドホルモンが妊娠の過程、特に妊娠期間や陣痛の開始にどのように影響を及ぼすかを理解する上で重要な洞察を提供します。
Moranら(2006年)は、21-OH欠損型非古典的CAH(NCAH)を持つ母親から生まれた子供のCAHおよびNCAHの頻度について研究しました。この研究では、21-OH欠損型NCAHの母親がCAHに罹患した子を出産するリスクが2.5%であり、これらの母親から生まれた子供の14.8%がNCAHであることが明らかにされました。この結果は、遺伝的要因がCAHおよびNCAHの発症リスクにどのように影響を及ぼすかを理解する上で貴重な情報を提供しています。
これらの研究は、CAHおよびNCAHに関連する遺伝的、生物学的、心理的要因を多角的に理解するための基盤を築いており、これらの疾患の管理や治療において考慮すべき重要な要素を示しています。
その他の特徴
マッピング
Dupontら(1977)は、21-ヒドロキシラーゼ欠損症とHLA複合体との密接な連鎖を示しました。Murtazaら(1978)はこの遺伝子とHLAとの連鎖を示唆する遺伝子を同定し、Levineら(1978)はその連鎖を強く支持するlodスコアを報告しました。Kloudaら(1980)はHLA-Bと21-ヒドロキシラーゼ欠損症との連鎖を見出し、Fleischnickら(1983)は拡張MHCハプロタイプが21-ヒドロキシラーゼ欠損症を引き起こす異なる変異のマーカーであることを証明しました。
これらの研究は、21-ヒドロキシラーゼ欠損症が特定のHLAハプロタイプと関連していることを示しており、HLAハプロタイプがこの遺伝性疾患の発症において重要な役割を果たしていることを示唆しています。また、特定のHLAハプロタイプを持つ人々が、21-ヒドロキシラーゼ欠損症の特定の型を発症するリスクが高いことも示しています。この知見は、21-ヒドロキシラーゼ欠損症の疾患機序の理解と、リスク評価および早期診断に役立つ可能性があります。
21-水酸化酵素欠損症は、遺伝的背景と強い関連性を持ち、特にHLA系との連鎖が認められています。この疾患の一般的な重症型はHLA-Bw47と正の相関を示し、HLA-B8とは負の相関を示す一方、遅発型はHLA-B14と正の相関を示します。HLAハプロタイピングは、21-水酸化酵素欠損症の遺伝的複雑性を明らかにするために使用されています。
特に、HLA-Bw47抗原を持つ患者は、21-ヒドロキシラーゼ活性とC4のC4A(ロジャース)型の同時欠損を示します。この関連性は、21-ヒドロキシラーゼ遺伝子座における欠失や再配列が、HLA-B13遺伝子と密接に関連していることを示唆しています。21-ヒドロキシラーゼ欠損症患者のDNA解析からは、特定のDNAバンドの欠失が確認され、この遺伝子座における変異の証拠が提供されました。
21-ヒドロキシラーゼ遺伝子の一つだけが変異している可能性があり、その機能はレニン-アンジオテンシン系によって制御され、特定の状況でのみ発現するか、あるいは偽遺伝子である可能性も考えられます。HLA-Bw47に関連するC4アロタイプの再検討からは、21-ヒドロキシラーゼ遺伝子がC4B(Chido)遺伝子の近くに位置し、特定のHLAハプロタイプでは両遺伝子が欠失する可能性があると結論付けられました。
さらに、21-ヒドロキシラーゼ欠損症の頻度に関する調査からは、非古典型遺伝子がHLA-B14と連鎖不平衡にあり、古典型がHLA-Bw47;DR7と連鎖不平衡にあることが示されました。遺伝的距離を推定する現在の方法では、21-ヒドロキシラーゼ遺伝子がHLA-Bに近いのかHLA-DRに近いのかについては、決定的な結論を得ることは困難です。
このように、21-水酸化酵素欠損症の遺伝的背景は複雑であり、特定のHLA型との関連性が疾患の異なる形態の理解に貢献しています。
遺伝
Spiroらによる1999年の研究では、常染色体劣性遺伝による突然変異のホモ接合体(またはコンパウンドヘテロ接合体)の減少が原因で発生した先天性副腎過形成の症例において、6番染色体の母親由来の片親ダイソミー(一方の親から受け継いだ染色体のコピーが2つある状態)の最初の事例を報告しました。このケースでは、母親はI172N変異(参照番号613815.0001)のヘテロ接合体であり、父親には検出可能な変異は見られませんでした。DNAのマイクロサテライト解析(特定の遺伝子領域の長さの変異を検出する方法)を用いて、6番染色体上の多型マーカーを調べた結果、患者からは母方の対立遺伝子のみが検出され、父方からは対立遺伝子が検出されなかったことが示されました。この患者は、子宮内での発育遅延が認められた後、出生してからキャッチアップ成長を遂げました。
頻度
原因
診断
出生前診断において、Gueuxら(1988年)は、HLAタイピングと連鎖解析を用い、罹患妊娠の羊水中で特定のステロイドの有意な上昇を認めました。Hughesら(1987年)は、21-ヒドロキシラーゼ欠損症の子供を持つ妊婦から採取した羊水の17-OHプロゲステロン濃度を測定し、塩喪失型CAHのみに信頼できる先天性副腎過形成の出生前診断が可能であると結論付けました。
より最近の技術として、Wudyら(1999年)は、妊娠中期の羊水中の複数のステロイドをプロファイリングすることで、21-ヒドロキシラーゼ欠損症の出生前診断において17-ヒドロキシプロゲステロンとアンドロステンジオンが最も有用であることを発見しました。
新生児の確定診断に関して、Caulfieldら(2002年)は、新生児期のCAHの正確で迅速、非侵襲的な鑑別診断を可能にするガスクロマトグラフィー/質量分析法を開発しました。また、Newら(1983年)は、21-水酸化酵素欠損症の軽症型および無症候性非古典型を区別するためのノモグラムを発表しました。
さらに、Bachegaら(2002年)は、CYP21遺伝子変異の保因者のACTH刺激後の17OHPレベルが、非古典型の診断において考慮すべき重要な要素であることを示しました。
最後に、Mornetら(1986年)は、HLA-DNAプローブの連鎖を利用した絨毛絨毛検体による妊娠初期の診断と、羊水中の17-ヒドロキシプロゲステロンの測定を組み合わせた方法を提案しました。
Reindollarら(1988年): 出生前診断に21-ヒドロキシラーゼ遺伝子のRFLP(制限断片長多型)を使用した初期の報告です。これは、遺伝的変異の特定における早期の方法の一つでした。
Leeら(1996年): CYP21遺伝子と非機能性CYP21P遺伝子の区別を可能にするPCRプライマーの開発と、CAHに関連する11の変異を特定するための二次PCR法を用いた。この方法により、出生前診断における変異の特定が簡便かつ直接的になりました。
Dayら(1996年): PCR増幅時の一つのハプロタイプの脱落が、無症候性の個体を誤ってタイピングする原因になることを示唆。彼らは、出生前診断において、マイクロサテライト型分類を用いることを推奨しました。
Lakoら(1999年): CAHの高精度の出生前診断を可能にする新しいマイクロサテライトマーカーを用いた連鎖分析法を開発しました。
Nordenstromら(1999年): 新生児スクリーニングを補完する診断ツールとして遺伝子型判定の価値を評価し、遺伝子型に応じて17-ヒドロキシプロゲステロンのレベルが異なることを発見しました。
Koppensら(2002年): CYP21A2遺伝子の変異解析を複雑にする重複について指摘し、正確な変異解析のためにハプロタイピングの重要性を強調しました。
Minuttiら(2004年)およびJanzenら(2007年): 新生児スクリーニングの偽陽性を減少させるために、液体クロマトグラフィー-タンデム質量分析を用いた第二段階の検査方法を提案しました。
Van der Kampら(2005年): CAHの新生児スクリーニングにおいて、17OHPのカットオフ値を決定する際に妊娠月齢を考慮することの重要性を指摘しました。
これらの研究は、CAHの診断に関する技術的進歩や、遺伝子型と臨床表現型の関連性を明らかにする上での課題を示しています。遺伝子診断技術の発展は、より正確で迅速な診断を可能にし、特に新生児スクリーニングや出生前診断における誤診のリスクを減少させることに寄与しています。
治療・臨床管理
Jones(1978年)は、成人で見られる軽度のCAHの症例を報告し、乏月経を示す患者に副腎皮質ステロイドでの治療を施しました。これは、通常のCAH治療法と同様のアプローチです。
Cutfieldら(1983年)は、部分的21-ヒドロキシラーゼ欠損症を持ち、両側精巣腫瘍と不妊症を呈した2人の従兄弟男性に対し、夜間の低用量デキサメタゾン療法によって腫瘍を消失させ、受胎可能性を回復させることに成功しました。
CutlerとLaue(1990年)は、ヒドロコルチゾン、抗アンドロゲン療法、アロマターゼ阻害剤を組み合わせた新しい治療法を提案しました。この治療法は、アンドロゲンの影響を抑え、正常な成長を目指すものです。
Forestら(1989年)によるフランスでの多施設共同研究では、21-ヒドロキシラーゼ欠損症の出生前治療として、母体にデキサメタゾンを経口投与する方法が検討されました。
Corneanら(1998年)は、21-ヒドロキシラーゼ欠損症の思春期前の小児22人を対象に、ステロイド治療が線状成長と骨格成熟に及ぼす影響を研究し、治療によって体脂肪が増加し、BMIが有意に上昇することを報告しました。
Nordenstromら(1999年)は、酢酸コルチゾンに対する反応がなかった21-ヒドロキシラーゼ欠損症の患者について報告し、この患者にはヒドロコルチゾンがより適切な代用療法であることを示唆しました。
TravitzとMetzger(1999)は古典的な21-水酸化酵素欠損症(21-OH型先天性副腎過形成)の出生前治療について議論しました。デキサメタゾン(DEX)は、その強力なグルココルチコイド作用、胎盤通過能、長い半減期、およびACTHに対する抑制効果の大きさから、出生前治療に適しているとされています。出生前DEX療法は賛否両論があります。
Loら(1999)は、古典的21-OH欠損症の女性4人における妊娠転帰と母体血清ステロイド濃度の連続測定を報告しました。これらの女性はDEX治療を受け、正常な女性外生殖器を持つ健康な女児を出産しました。妊娠中のアンドロゲンレベルは上昇しましたが、DEX治療中は正常範囲内に保たれました。胎盤のアロマターゼ活性が胎児の外性器と脳の男性化を防ぐのに十分であったとされています。
Laueら(1996)は、フルタミド、テストラクトン、ヒドロコルチゾンの減量、フルドロコルチゾンを含む4剤併用治療レジメンが、線状成長、体重増加、骨の成熟をより良好にコントロールしたことを報告しました。Merkeら(2000)はこの治療レジメンが、グルココルチコイド過剰のリスクを減らしながらCAHを効果的にコントロールできることを示しました。
Charmandariら(2001)は、思春期における21-水酸化酵素欠損症の患者の治療とコルチゾールの薬物動態の変化を調査し、思春期においてコルチゾールのクリアランスと分布量が増加し、遊離コルチゾールの半減期が性別によって異なることを発見しました。これらの変化は、思春期におけるCAHの治療に影響を与える可能性があるとされています。
塩類消耗性21-OHD CAH患者54人に関するMuirheadら(2002年)の研究では、成人身長が乳児期および小児期のアンドロステンジオンおよび小児期のテストステロンと負の相関があることが見られました。これは、21-OHD CAH患者において、グルココルチコイド治療の最適化が重要であることを示唆しています。
Bonfigら(2007年)は、CAH患者125人における最終身長とステロイド治療の影響を研究し、従来の治療で十分な最終身長を達成できると結論付けましたが、思春期の成長は有意に低下し、プレドニゾンによる治療は最終身長を低下させると報告しました。
LWPES/ESPE合同CAHワーキンググループは、21-ヒドロキシラーゼ欠損症によるCAHの管理に関するベストプラクティスとガイドラインを提供しましたが、Creightonら(2003年)は、外科的管理に関するガイドラインに異議を唱え、個々の症例に対する学際的な専門チームによるアプローチの重要性を強調しました。
Van WykとRitzen(2003年)は、両側副腎摘出術を受けたCAH患者18人の研究を要約し、この手術が選択された患者にとって安全かつ有効であると結論付けました。
デキサメタゾンの出生前投与は、CAHの女性胎児の生殖器男性化予防に用いられますが、Meyer-Bahlburgら(2004年)の研究では、この治療が運動発達や認知発達に悪影響を与える証拠は見つかりませんでした。しかし、Hirvikoskiら(2007年)は、デキサメタゾン投与が言語性ワーキングメモリーに長期的な悪影響を及ぼす可能性があると報告しました。
Sciannambloら(2006年)およびFalhammarら(2007年)は、CAH患者において骨密度が低下し、骨折リスクが高いことを示し、早期からの骨密度モニタリングと適切な予防と治療の重要性を強調しました。
Nordenskjoldら(2008年)は、CAH女性の女性化手術の結果について調査し、手術方法と遺伝子型が性生活および全般的なQOLに影響を及ぼすことを発見しました。
これらの研究は、CAHの治療と管理における複数の側面に光を当てており、個々の患者に対する包括的で個別化されたアプローチの重要性を強調しています。
分子遺伝学
21-ヒドロキシラーゼ欠損症の臨床的表現は非常に多岐にわたり、疾患の重症度は遺伝子変異のタイプや位置によって異なります。最も重篤な形態は、塩類喪失型CAHであり、新生児期に重篤な電解質異常を引き起こす可能性があります。非塩類喪失型(簡易型)CAHは比較的軽度であり、通常は思春期や成人期に診断されます。
CYP21A2遺伝子の変異は多種多様で、大きな欠失や微小挿入、点変異が報告されています。これらの変異は、遺伝子の機能を損ない、21-ヒドロキシラーゼ酵素の活性を低下させることで疾患を引き起こします。
この疾患の分子遺伝学に関するさらに詳細な情報は、こちらのページを参照してください。ここでは、遺伝子変異の特定、変異が酵素活性に与える影響、疾患の遺伝的異質性についての包括的な情報が提供されています。これらの知見は、診断、治療選択、および遺伝カウンセリングにおいて重要な役割を果たします。
遺伝子型と表現型の関係
一方、Nikoshkovら(1997)は、遅発性CAHを持つ2人の兄弟姉妹におけるまれな対立遺伝子を研究し、3つの配列変化(-4C-to-T転移、pro105-to-leu置換、pro453-to-ser置換)を含むアレルを特定しました。これらの変異のうち、pro105-to-leuとpro453-to-serの変異はそれぞれ酵素活性を著しく低下させ、両方の変異が組み合わさることでさらに低下しました。これらの結果は、これらの変異が非常に微妙な疾患を引き起こす可能性があるが、組み合わされた場合には軽症のCAH患者の遺伝子型決定に重要であることを示唆しています。
Witchelら(1996年)は、対立遺伝子特異的オリゴヌクレオチドハイブリダイゼーション、SSCP、ヘテロ二重鎖分析を用いて21家族38人のCYP21遺伝子変異を同定しました。これらの個体はすべて、21-水酸化酵素欠損症でよく見られるイントロン2のスプライシング変異のホモ接合体または複合ヘテロ接合体でした。彼らの研究は、古典的な塩類喪失型21-ヒドロキシラーゼ欠損症から無症状の表現型まで、表現型の不均一性を示しました。この表現型の異質性について、彼らはいくつかの可能性を示唆しました。
Miller(1997年)は、21-ヒドロキシラーゼ欠損症の表現型の不均一性を説明するために、ステロイド21-ヒドロキシラーゼ活性を持つ他のタンパク質をコードする遺伝子の活性の可能性を指摘しました。これは、チトクロームP450酵素が多種多様な水酸化反応を触媒する「乱交性」酵素であるという特性に基づいています。
LevoとPartanen(1997年)は、フィンランドの51の非血縁家系におけるCYP21遺伝子の突然変異と組換えブレイクポイントを解析し、この集団で複数の創始者変異とハプロタイプの組み合わせが存在することを発見しました。この研究は、遺伝的多様性と創始者効果が21-水酸化酵素欠損症の遺伝学において重要な役割を果たしていることを示しています。
Jaaskelainenら(1997年)は、フィンランドの21-水酸化酵素欠損症患者120人の集団を解析し、遺伝子型と表現型の間に一般的に良好な相関があることを発見しました。特に、重篤な酵素活性の影響を及ぼす変異を持つ患者は、塩類消耗型の21-ヒドロキシラーゼ欠損症であることが確認されました。
Wedell(1998年)は、スウェーデンでの21-ヒドロキシラーゼ欠損症の診断において、1990年以降、直接変異検出が行われてきたことを報告しました。分析された約400の21-水酸化酵素遺伝子の大部分は偽遺伝子との相互作用による突然変異であり、これには遺伝子欠失と9つの小さな配列異常が含まれていました。これらの変異は、重症度に応じて分類され、遺伝子型に基づいて罹患者の臨床転帰を予測することが可能になりました。
Dacou-Voutetakis and Dracopoulou(1999年)は、早発性副腎皮質機能不全(PA)児のCYP21遺伝子を解析し、一部の被験者で臨床的に表現されていることを発見しました。彼らは、これらの小児を思春期まで追跡調査する必要性を強調しました。
Bachegaら(1998年)は、ブラジルの古典型および非古典型CAH患者における点突然変異の頻度を測定し、遺伝子型と表現型との相関を調査しました。最も頻度の高い変異は、塩類消耗型、単純男性型、遅発型に関連するものでした。彼らの研究は、患者の臨床転帰を遺伝子型に基づいて予測することの重要性を示しています。
Nimkarnら(1999年): CAH1患者とその家族のCYP21遺伝子を解析し、3.7kbの配列に変異は認められなかったと報告し、CYP11B1遺伝子の欠損がCAH1の臨床的および生化学的表現型に酷似する可能性のあることを除外しました。
Kroneら(2000年): 155人の血縁関係のないCAH患者を対象に変異の頻度と遺伝子型と表現型の関係を調査し、遺伝子型と臨床的表現型との間に一般的に良好な相関があることを示しましたが、中程度の重症度の変異群では相違が観察されました。
Dracopoulou-Vabouliら(2001年): ギリシャ人集団におけるCYP21遺伝子の変異とその頻度、遺伝子型と表現型の相関を調査しました。特定の変異は特定の表現型と関連していましたが、遺伝子型と表現型の一致率は疾患の重症度が低くなるにつれて低下することが示されました。
Deneuxら(2001年)は、症候性非クラスの先天性副腎過形成(CAH)を有するフランス人女性56人を対象にCYP21遺伝子の解析を行い、変異スペクトルと表現型-遺伝子型の相関を検討しました。この研究で最も一般的に見られた変異は、val281からleuへの変異で、対象女性の80%に見られ、対立遺伝子の51%に存在しました。また、3つの新規変異が発見されました。63%の女性がCYP21遺伝子に重篤な変異を有しており、これにより彼女たちは古典的な型の子供を出産する危険性があるとされました。著者らは、遺伝子型から表現型を正確に予測することは難しいと結論づけ、表現型の多様性はCYP21遺伝子座の遺伝的不均一性以外のメカニズムによっても調整されている可能性があると指摘しました。
L’Allemandら(2000年)は、新生児スクリーニングで中等度に17-ヒドロキシプロゲステロン値が上昇した非クラスの21-ヒドロキシラーゼ欠損症の症例を報告しました。この女性患者は男性化の徴候は示さず、コルチゾール、プラズマレニン活性(PRA)、電解質は正常でしたが、17-ヒドロキシプロゲステロンと21-デソキシコルチゾールの濃度は上昇していました。サザンブロット法によると、この患者はCYP21P偽遺伝子の3-プライムエンド、C4B遺伝子、機能的CYP21遺伝子の5-プライムエンドを含む30-kb欠失のホモ接合体であり、さらに8bp欠失のヘテロ接合体でもあることが示されました。このケースは、古典的CAHと非典型的CAHの中間型の可能性を示唆しており、特定の遺伝子型が非常に珍しい表現型を引き起こす例として注目されています。
Tusie-LunaとWhite(1995)によって、CYP21PとCYP21遺伝子の5-プライム末端がCYP21Pに、3-プライム末端がCYP21に対応するキメラ遺伝子が同定され、これがフレームシフトとトランケートタンパク質を生じるdeleterious mutationにより非機能性であることが明らかにされました。Leeら(2002)は、CAH患者における2つのキメラCYP21P/CYP21遺伝子を報告し、これらが不等間隔クロスオーバーの結果として生じたことを示しました。
Charmandariら(2002)は、塩類消耗性CAHと単純性男性化型CAHの患者を対象に、副腎髄質機能、疾患の重症度、遺伝子型との関連を検討しました。彼らは遺伝子型と表現型の不一致があり、血漿中の遊離メタネフリン濃度が疾患の重症度を予測する有用なバイオマーカーであることを示しました。
SpeiserとWhite(2003)は、CYP21の突然変異を3つのカテゴリーに分類し、これらが表現型に与える影響について議論しました。彼らは、遺伝子型-表現型のばらつきの一因としてスプライス変異の漏出性を指摘しました。
これらの研究は、CAHの診断と治療における遺伝子型と表現型の複雑な関係を浮き彫りにし、特定の遺伝子変異が疾患の臨床的特徴にどのように影響するかを理解するための基盤を提供しています。これは、個々の患者に最適な治療戦略を選択する際に重要な考慮事項となります。
集団遺伝学
スイスのチューリッヒ州では、Prader(1958)によって、先天性副腎過形成の頻度は出生5,041人に1人と推定され、保因者の頻度は35人に1人とされています。これに対して、メリーランド州ではChildsら(1956)による推定では67,000出生に1人と、はるかに低い頻度で発症していることが示されています。
トロントでは、QaziとThompson(1972)による食塩喪失性C-21水酸化酵素欠損症の最小頻度は26,292人に1人と推定されています。また、アラスカのエスキモーでは、Hirschfeld and Fleshman(1969)によって、21水酸化酵素欠損症の塩類喪失型が比較的高頻度で見られることが示されています。
非典型性21-ヒドロキシラーゼ欠損症は、特にアシュケナジム人、ヒスパニック系、ユーゴスラビア人、イタリア人の間で高頻度であり、Speiserら(1985)によって最も頻度の高い常染色体劣性遺伝病の一つであると結論づけられています。この遺伝子は特定のHLAハプロタイプとの連鎖不平衡を示し、古典型CAHはHLA-Bw47;DR7と連鎖不平衡を示すとされています。
これらのデータは、先天性副腎過形成が地域や民族集団によって異なる遺伝的背景を持つ複雑な疾患であることを示しています。特定の集団における高頻度は、遺伝的スクリーニングや集団特有のリスク管理戦略の重要性を強調しています。
Layrisseら(1987年)は、塩類消耗型21-水酸化酵素欠損症を持つ新生児20人を含む混血のベネズエラ人19家族について調査し、HLAハプロタイプとコンプロタイプを決定しました。彼らの研究結果は、一般的な報告とは異なり、HLA-Bw47や拡張ハプロタイプHLA-Bw47,DR7,FC91,0との関連が観察されませんでした。この研究は、塩類消耗型21-水酸化酵素欠損症が比較的少数の独立した突然変異による創始者効果から主に生じる可能性を示唆しています。
ThilenとLarsson(1990年)は、1969年から1986年の間にスウェーデンで生まれた全CAH患者を対象に新生児スクリーニングの有益性を検討するレトロスペクティブ研究を実施し、古典型CAHが143人、非典型的CAHが7人であることを確認しました。塩分喪失は93例に認められ、特に女児で性別の割り当てが問題となることが多く見られました。
Chrousosら(1982年)は、多毛症の女性の6〜12%が軽度の21-水酸化酵素欠損症であると推定し、この減弱型遺伝子の頻度を0.015から0.057と計算しました。
日本では、1991年から1994年にかけて約450万人の乳児がCAHの新生児スクリーニングを受け、非典型的ステロイド21水酸化酵素欠損症の可能性が高い4症例が同定されました。これらの症例は特定の変異を持つ遺伝子変換によって特徴づけられていました。新生児スクリーニングによる非典型型の推定検出率は、古典型に比べて低いことが示唆されています。
これらの研究結果からは、21-水酸化酵素欠損症の臨床的および遺伝的多様性が浮き彫りになり、特に新生児スクリーニングの有益性と限界が示されています。また、疾患の遺伝的背景に関する深い理解が、より効果的な診断と管理戦略の開発に繋がる可能性があります。
Witchelら(1997年)は、21-水酸化酵素欠損症のヘテロ接合体が生存に有利である可能性があるという仮説を立て、証明された保因者においてコルチゾール反応が高いことを発見しました。これは感染性や炎症性の環境ストレスに迅速に対応し、不適切な免疫応答から保護する効果があると提唱されました。
Wedell(1998年)は、スウェーデンで解析された約400の21-水酸化酵素遺伝子から、9つの一般的な偽遺伝子由来の突然変異が対立遺伝子の約95%を占めていることを報告しました。この知見により、罹患者の臨床転帰を遺伝子型に基づいて予測することが可能になりました。
Lakoら(1999年)は、英国人集団におけるCYP21突然変異のスクリーニングを報告し、最も一般的な変異が大規模欠失または転換、イントロン2スプライス変異、R357W、I172Nであることを示しました。
Ferencziら(1999年)は、ハンガリーのCAH患者におけるイントロン2のスプライス変異が最も頻度の高い変異であることを報告し、遺伝子型と表現型の相関が良好であることを発見しました。
New and Wilson(1999年)は、21-OH欠損症を引き起こすCYP21遺伝子の約40個の変異が同定され、最も一般的な変異はCYP21と偽遺伝子CYP21Pの間の減数分裂相互作用の結果であると述べました。
Fitnessら(1999年)は、新生児スクリーニング検体からCYP21変異の分析が可能であり、古典的および非古典的CYP21変異のヘテロ接合体率をニュージーランド人集団でそれぞれ2.8%および2.0%であることを示しました。
Baumgartner-Parzerら(2005年)は、中欧(オーストリア)の集団におけるCAH保因者頻度が9.5%であることを発見し、これは以前推定されていたよりも高い有病率を示していると結論づけました。
これらの研究は、21-ヒドロキシラーゼ欠損症に関する深い理解をもたらし、遺伝的変異の同定、遺伝子型と表現型の相関、保因者の検出方法など、疾患の診断と管理における重要な進展を示しています。
Wilsonら(2007年)の研究は、21-ヒドロキシラーゼ欠損症(CAH)患者716人におけるCYP21A2遺伝子変異の民族特異的分布を明らかにしました。この研究では、変異と遺伝子型において民族間で有意な差が存在することが確認され、特定の変異が特定の民族集団で顕著に多いことが示されました。例えば、大きな欠失変異はアングロサクソンに、V281L変異はアシュケナージ・ユダヤ人に、R356W変異はクロアチア人に、IVS2AS-13変異はイラン人と西アラスカのユピック語を話すエスキモー人に、Q318X変異は東インド人に多くみられました。また、IV2AS-13変異が存在する場合に遺伝子型と表現型の非相関が観察されたことも興味深い所見です。
一方、Hannah-Shmouniら(2017年)は、アシュケナージ・ユダヤ人と無作為に選ばれた米国白人を対象に非古典型CAHの保因者率を調査しました。その結果、アシュケナージ・ユダヤ人と白人の間で非古典型CAHの保因者率に有意な差はなかったものの、アシュケナージ・ユダヤ人における非典型型CAHの保因者及び罹患者の割合は、過去の報告ほど高くはなかったことが示されました。また、白人における非古典型CAHの推定有病率は200人に1人(0.5%)であり、非典型型CAHは民族に関係なく一般的な疾患であると結論付けられました。
Pintoら(2003年)の研究では、21-水酸化酵素欠損症(21-OHD)による先天性副腎過形成(CAH)患者68人を対象に、臨床的、ホルモン学的、分子遺伝学的特徴を評価しました。患者は遺伝子型に基づき分類され、その結果、0群(ヌル変異)とA群(重度の変異)は全員が塩類消耗型であり、C群は非古典型、B群には塩類消耗型と単純童貞型の両方が含まれていました。診断時の年齢が若く、女性の処女化が進んでいることが、0群およびA群の特徴であることが示されました。著者らは、遺伝的欠陥の重症度と臨床的特徴の間には強い相関があると結論付け、新生児スクリーニングや遺伝子型判定を組み合わせた治療がCAHの管理に有用であると述べています。
Stikkelbroeckら(2003年)の研究では、オランダの198人の21-ヒドロキシラーゼ欠損症患者におけるCYP21遺伝子変異の頻度と遺伝子型と表現型の相関を調査しました。最も一般的な変異はI2GとI172Nであり、特定の変異は特定の表現型と強く関連していることが示されました。オランダの患者集団において特有の変異パターンが見られ、新規変異も発見されました。
Soardiら(2008年)は、ブラジル人およびスカンジナビア人患者におけるCYP21A2遺伝子の新規および再発性変異の機能的影響を調査しました。H62L変異は非典型的変異の範囲内で活性を示し、変異の特定が患者の診断と治療の選択に役立つことを示しています。
これらの研究は、21-OHD CAHの診断と管理において、遺伝子型と表現型の綿密な比較が重要であることを示しています。特に、遺伝子型の特定は、治療計画の策定、患者のフォローアップ、および家族への遺伝相談に有用な情報を提供します。これにより、個々の患者に対するより適切な治療戦略の選択が可能となり、患者のQOLの向上につながる可能性があります。
動物モデル
また、Gotohらによる1988年の研究では、特定のH-2組み換えハプロタイプのマウスにおける21-ヒドロキシラーゼ遺伝子の欠失が説明されました。これらの新生児ホモ接合体マウスは21-ヒドロキシラーゼ活性の欠損症を持ち、生後早期に死亡することが見出されました。新生児ホモ接合体の副腎では形態学的変化が観察されています。これらの研究は、21-ヒドロキシラーゼ遺伝子の発現とその欠損が副腎機能に及ぼす影響を理解する上で重要な貢献をしています。
疾患の別名
CYTOCHROME P450, SUBFAMILY XXI; CYP21
STEROID CYTOCHROME P450 21-HYDROXYLASE; P450C21
21-HYDROXYLASE B; CYP21B
CA21H
CYTOCHROME P450, SUBFAMILY XXIA, POLYPEPTIDE 1 PSEUDOGENE, INCLUDED; CYP21A1P, INCLUDED
CYP21P, INCLUDED; CYP21A, INCLUDED







