疾患に関係する遺伝子/染色体領域
疾患概要
PIGMENTED PARAVENOUS CHORIORETINAL ATROPHY; PPCRA
色素性静脈傍脈絡膜網膜萎縮症(PPCRA)は、染色体1q31上のCRB1遺伝子(604210)のヘテロ接合体変異によって引き起こされる眼疾患です。このため、この項目には番号記号(#)が使用されています。CRB1遺伝子は、網膜の細胞間相互作用や細胞極性の維持に重要な役割を果たすタンパク質をコードしており、その変異は視覚に関連するさまざまな障害を引き起こす可能性があります。
PPCRAは、眼底における骨角膜色素沈着が静脈の周囲に特異的に分布することを特徴とする定常性疾患です。この疾患は通常、患者に明らかな症状を引き起こさないため、特徴的な眼底像に基づいて診断されます。ほとんどの症例は男性に報告されています。
PPCRAは、網膜における色素の異常な蓄積と関連していますが、多くの場合、視力に重大な影響を与えることはありません。しかし、CRB1遺伝子の変異によって引き起こされるため、他のCRB1関連疾患と共通の病理機序を共有する可能性があります。これにより、CRB1遺伝子の研究は、PPCRAだけでなく、他の視覚障害の理解と治療法の開発に貢献する可能性があります。
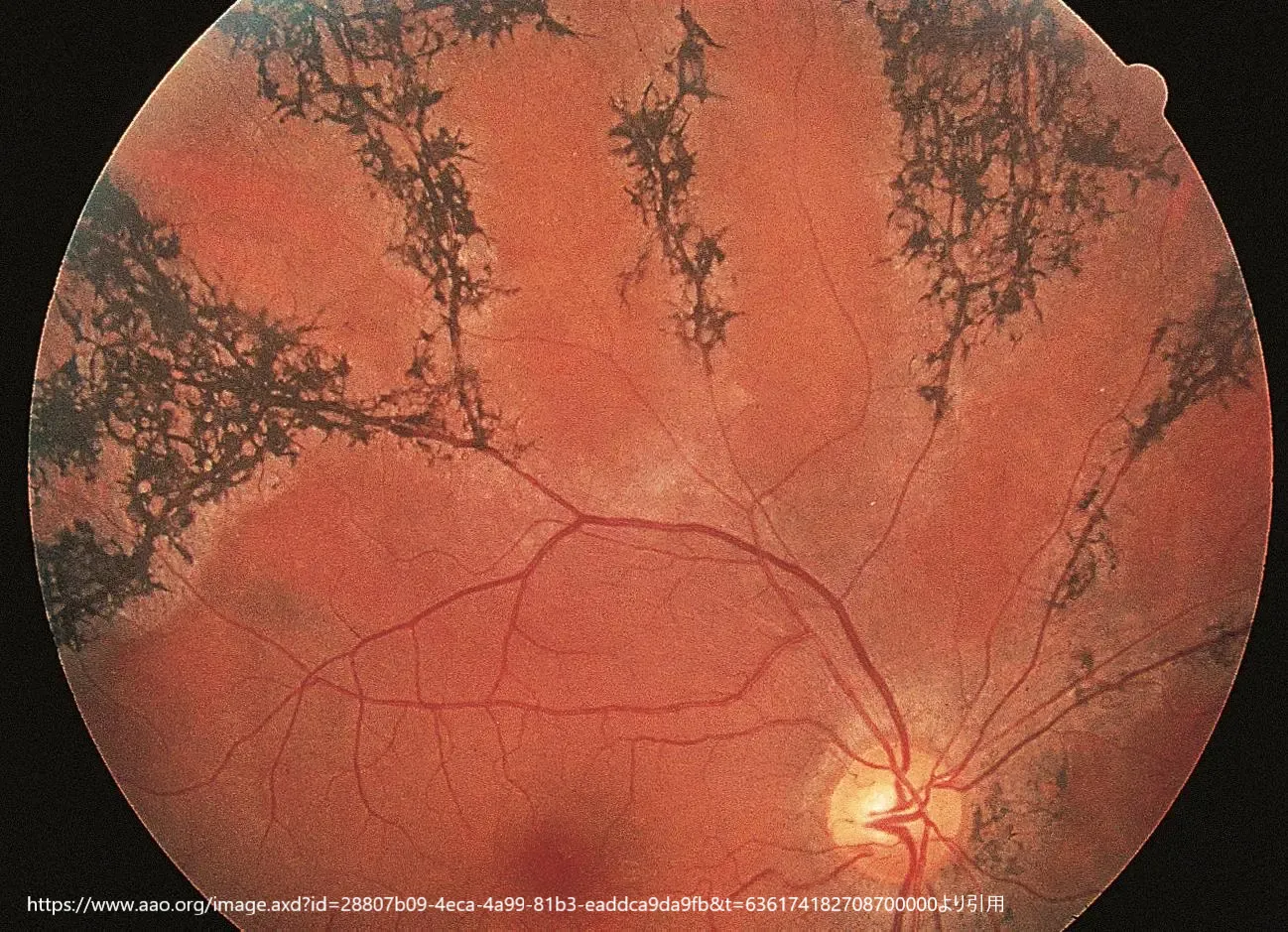
臨床的特徴
– Skalka(1979年)は、罹患した父と息子のケースを報告し、疾患が家族内で伝わる可能性を示唆しました。
– Traboulsi and Maumenee(1986年)は、母親と3人の息子がPPCRAを発症し、遠視や内斜視、網膜硝子体変性を伴う家族を報告しました。症状は母親では軽度、息子たちでは重度でした。
– Noble(1989年)は、非血縁の両親から生まれた3人の兄弟に傍大脳骨棘蓄積が見られるケースを報告し、疾患の進行が非常に緩慢であることを示しました。
– McKayら(2005年)は、PPCRAが優性遺伝する家族を報告しました。この家族では、症状の発現が様々で、男性が重度の表現型を示しやすい一方で、女性は晩年まで無症状である可能性があると述べられています。
– Choiら(2006年)による長期追跡研究では、PPCRA患者における周辺視力の低下が緩徐に進行することが示されました。
これらの研究は、PPCRAが多様な臨床的表現を持ち、進行速度が個々の患者で異なることを示しています。また、性別によっても症状の重さが異なる可能性があり、特に男性患者で重度の表現型が見られる傾向にあります。PPCRAの診断は、特徴的な眼底像に基づいて行われ、多くの場合、患者は無症状であるため、定期的な眼科検診が重要です。
遺伝
– Skalka(1979)による報告では、PPCRAは常染色体優性遺伝のパターンを示し、父親から子への伝達が観察されました。この遺伝の形式では、影響を受ける遺伝子の単一のコピーが疾患を引き起こすため、性別に関係なく疾患が発現します。
– 一方、Traboulsi and Maumenee(1986)が報告した血統では、疾患の表現型が男性でより重篤であったため、X連鎖遺伝と一致すると考えられました。X連鎖遺伝の場合、影響を受ける遺伝子がX染色体上に位置しており、男性はX染色体を1本しか持たないため、影響を受ける遺伝子の変異があると疾患が発現しやすくなります。
– McKayら(2005)の報告でも、PPCRAは優性遺伝であるとされていますが、この情報はSkalka(1979)の報告と一致しており、疾患が優性遺伝の特性を持つことを補強しています。
これらの異なる報告は、PPCRAに関連する遺伝メカニズムが複雑であること、そして特定の疾患や家系で異なる遺伝パターンが観察され得ることを示しています。遺伝的な背景の詳細な解明は、適切な診断、リスク評価、およびカウンセリングに不可欠です。さらなる研究によって、PPCRAを含む多くの遺伝性疾患の遺伝学的基盤が明らかになることが期待されています。
分子遺伝学
CRB1遺伝子は、網膜の細胞間相互作用や細胞の極性の維持に重要な役割を果たすタンパク質をコードしています。そのため、CRB1遺伝子の変異は、網膜ジストロフィーなどの視覚障害を引き起こす原因となることが知られています。特に、第4EGF様ドメインは、CRB1タンパク質の細胞外ドメインの一部であり、細胞外マトリックスとの結合や他の細胞表面タンパク質との相互作用に関与していると考えられています。
この研究は、特定のCRB1遺伝子変異が特定の家系における視力障害の原因であることを示し、CRB1関連疾患の遺伝的基盤に関する理解を深めました。また、これらの発見は、CRB1遺伝子変異を有する患者の診断や管理において有用な情報を提供し、将来的な治療法の開発に向けた重要な手がかりとなります。







