目次

家族性大腸腺腫症(FAP)は、APC遺伝子の生殖細胞系列変異によって若年期から大腸に数百〜数千個のポリープが発生する遺伝性疾患です。未治療では生涯ほぼ100%の確率で大腸がんを発症するため、早期の遺伝子診断と生涯にわたる計画的なサーベイランス、そして適切なタイミングでの予防的手術が患者さんの命を守る鍵となります。
Q. 家族性大腸腺腫症(FAP)とはどのような病気ですか?まず結論だけ知りたいです
A. APC遺伝子の生まれつきの変異により、10代から大腸に数百〜数千個のポリープが発生し、未治療では平均39歳で大腸がんを発症する常染色体顕性遺伝の疾患です。適切な内視鏡サーベイランスと予防的大腸切除によって大腸がん死を回避することは十分に可能であり、十二指腸がん・甲状腺がん・デスモイド腫瘍など全身の合併症管理も生涯にわたって重要です。
- ➤疾患の定義 → APC遺伝子変異・常染色体顕性遺伝・生涯発がんリスクほぼ100%
- ➤分子メカニズム → Wnt/β-カテニン経路の暴走と腺腫・がん連続過程
- ➤表現型スペクトラム → 古典型FAP・減弱型(AFAP)・GAPPS・ガードナー・ターコット症候群
- ➤サーベイランス → 大腸内視鏡10〜15歳開始・Spigelman分類による上部消化管管理
- ➤治療と最新研究 → IPAA/IRAの選択・化学予防・デスモイド治療薬ニロガセスタット(2023年FDA承認)
1. 家族性大腸腺腫症(FAP)とは
家族性大腸腺腫症(Familial Adenomatous Polyposis:FAP)は、APC遺伝子の生殖細胞系列変異によって引き起こされる、常染色体顕性遺伝形式の消化管ポリポーシス症候群です。患者さんの大腸には10代という若年期から無数の腺腫性ポリープが発生し、適切な医学的介入が行われない場合、生涯にわたってほぼ100%の確率で大腸がん(CRC)を発症します。
💡 用語解説:常染色体顕性遺伝(じょうせんしょくたいけんせいいでん)
「常染色体」とは性染色体(X・Y)以外の染色体のこと。「顕性(優性)」とは、対になっている2本の染色体のうちどちらか1本に変異があるだけで症状が現れる遺伝形式を指します。FAPでは親から子へ遺伝する確率は理論上50%です。性別に関係なく男女ともに同じ確率で受け継がれます。
💡 用語解説:腺腫(せんしゅ/adenoma)とポリポーシス
腺腫とは、消化管の粘膜から発生する良性のポリープの一種で、放置するとがんに進行する可能性のある「前がん病変」です。ポリポーシスとは消化管に多数(一般的に100個以上)のポリープが発生している状態を指します。FAPでは大腸全体に数百〜数千個もの腺腫性ポリープが発生します。
疫学:どのくらい稀な病気か
FAPの発生頻度に男女差はなく、有病率は7,000〜31,000人に1人、発生率としては約8,500人に1人と報告されています。すべての大腸がん症例全体に占めるFAPの割合は1%未満(0.5%未満とする報告もあり)と稀ですが、発症者の極めて高いがん化リスクから、公衆衛生および臨床腫瘍学において重要な疾患モデルとなっています。遺伝性大腸がん症候群のなかでは、リンチ症候群に次いで2番目に多い症候群です。
最新のコホート研究によれば、FAP患者は大腸がんを発症する確率が非FAP患者と比較して有意に高く、結腸がんのハザード比は2.16〜2.72、直腸がんに至ってはハザード比9.12〜12.66という驚異的な数値が報告されています。
FAPは「単一の病気」ではない——表現型スペクトラム
FAPは単一の疾患単位としてではなく、変異の生じる位置や種類によって劇的に異なる臨床像を示すスペクトラム障害として理解されるべき疾患です。主な表現型として以下があります。
🔴 古典型FAP(Classic FAP)
大腸に100個以上のポリープ。平均16歳から発症、35歳までに95%にポリープ多発。未治療では平均39歳で大腸がん発症。
🟡 減弱型FAP(AFAP)
ポリープは100個未満で右側結腸寄り。発症が遅く大腸がん発症は平均50〜55歳。それでも生涯発がんリスクは約70%。
🔵 GAPPS
APCプロモーター1B変異により、胃の近位部だけに絨毯状のポリポーシスが発生する特殊型。胃腺がんリスクが極めて高い。
🟢 ガードナー/ターコット症候群
FAPに骨腫・デスモイド腫瘍・脳腫瘍などの大腸外所見を合併する病型。同じAPC変異から多彩な病態が生じる。
🔍 関連記事:FAPの各サブタイプを詳しく知りたい方は 減弱型FAP(AFAP)/GAPPS/ガードナー症候群/ターコット症候群 をご覧ください。
2. なぜ大腸にポリープが多発するのか:分子メカニズム
FAPの病態を理解するうえで核心となるのが、APC遺伝子と、その産物であるAPCタンパク質が制御するWnt(ウィント)/β-カテニン・シグナル伝達経路です。この経路の暴走こそが、大腸粘膜の異常増殖を引き起こす根本的な原因です。
💡 用語解説:APC遺伝子
APC(Adenomatous Polyposis Coli)遺伝子は、第5番染色体長腕(5q22.2)に位置する代表的ながん抑制遺伝子です。15個の配列エクソンと2,843個のアミノ酸(コドン)からなる巨大な遺伝子で、Wntシグナル経路の制御・細胞接着・細胞遊走・染色体安定性など多彩な役割を担います。FAP患者で見られる変異の大部分は遺伝子の5’側半分に集中し、特にコドン1061と1309周辺がホットスポットとして知られています。
💡 用語解説:がん抑制遺伝子(Tumor Suppressor Gene)
細胞の異常な増殖にブレーキをかけ、がん化を防ぐ役割を持つ遺伝子の総称です。「車のブレーキ」にたとえられ、両方のアレル(父由来・母由来の2本の遺伝子コピー)が壊れて初めて機能不全に陥り、がん化が起こります。これを「ツーヒット仮説(Knudson仮説)」と呼びます。FAPではすでに片方のアレルに生まれつきの変異(ファーストヒット)があるため、もう片方が後天的に壊れる(セカンドヒット)と直ちに腺腫が発生します。
破壊複合体の崩壊がポリープを生む
正常な細胞ではAPCタンパク質が、Axin・GSK-3βなどの分子と組み合わさって「破壊複合体(Destruction Complex)」を形成しています。この複合体は、細胞内で増えすぎたβ-カテニンを捕まえてリン酸化し、ユビキチン・プロテアソーム系によって分解へと誘導します。これにより細胞増殖が適切に抑制されています。
しかしFAP患者で生じるAPC変異の多くは、機能的に不完全な「短縮型APCタンパク質」を産生します。短縮型APCはWntシグナル制御や細胞骨格との相互作用に必要なC末端領域を欠いているため、破壊複合体が機能不全に陥り、β-カテニンを分解できなくなります。
💡 用語解説:β-カテニンとWntシグナル経路
β-カテニンは細胞増殖や分化を制御する重要な転写因子です。通常は破壊複合体によって速やかに分解されていますが、APCが壊れると分解されずに細胞質に蓄積し、核内に移行します。核内でβ-カテニンはTCF/LEFという転写因子と結合し、c-MycやCyclin D1などの細胞増殖遺伝子を恒常的にオンにしてしまいます。これが大腸粘膜での無秩序な細胞増殖、すなわち腺腫形成の引き金です。
腺腫・がん連続過程(アデノーマ・カルシノーマ・シークエンス)
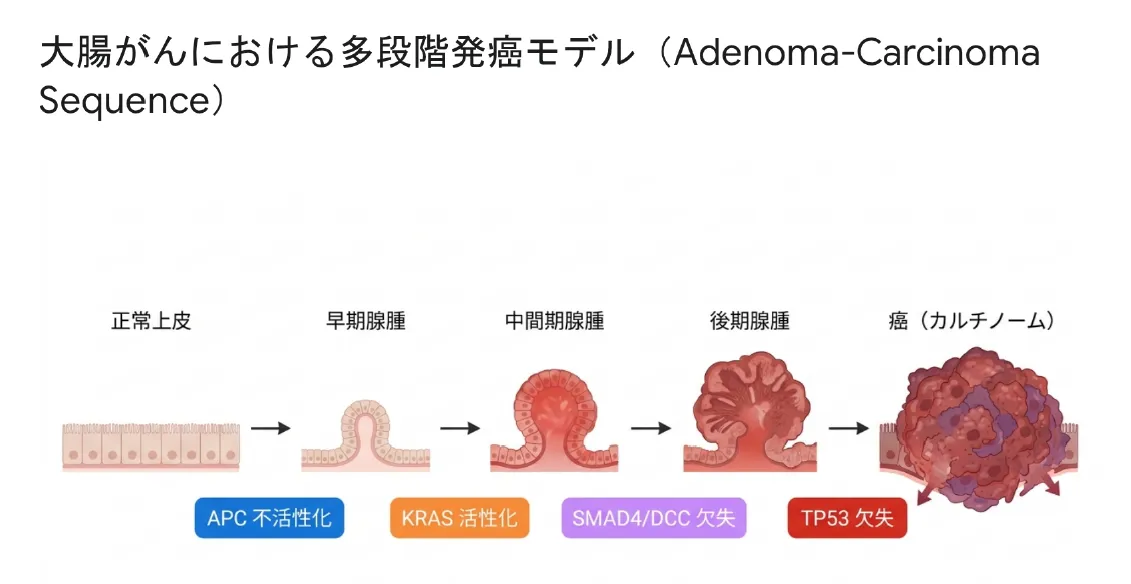
正常大腸粘膜はAPC変異(ファースト/セカンドヒット)によって最初に腺腫を形成し、その後KRAS変異・p53変異などが段階的に蓄積することで、小腺腫→大腺腫→高度異形成→浸潤がんへと不可逆的に進行する。
APCの不活性化は、大腸発がんにおける「最初のスイッチ」に過ぎません。腺腫が形成された後、KRAS変異・p53変異など他の発がん遺伝子・がん抑制遺伝子の変異が段階的に蓄積することで、組織は正常粘膜→小腺腫→大腺腫→高度異形成→浸潤がんへと不可逆的に進行していきます。これが大腸がん発生のスタンダードモデルとされる「アデノーマ・カルシノーマ・シークエンス(腺腫・がん連続過程)」です。
変異の場所が病気の重さを決める:遺伝子型・表現型相関
FAPの臨床像は、APC遺伝子のどこに変異があるかによって強く規定されます。これを「遺伝子型・表現型相関(Genotype-Phenotype Correlation)」と呼びます。
・コドン1250〜1464の変異 → 重症型(数千個のポリープが密集する古典型)
・遺伝子の極端な5’末端や3’末端の変異 → 減弱型FAP(AFAP)
・APCプロモーター1Bの点変異 → GAPPS(胃近位部のみのポリポーシス)
・3’側変異 → ガードナー症候群(デスモイド腫瘍・骨腫を合併しやすい)
より詳細にコドン領域ごとの相関を整理すると、次の表のようになります。この相関は、個々の患者さんに対するオーダーメイドのサーベイランス計画を構築するうえで不可欠な情報です。
特筆すべきは、デスモイド腫瘍リスクが変異コドン位置に極めて鋭敏に反応する点です。コドン400より5’側では発生率14.9%であるのに対し、コドン1400より3’側では37.1%に達します。ただし遺伝子型は表現型の「絶対的予測因子」ではなく、AFAP関連領域の変異を持つ方でも約35%が最終的に古典型FAPの表現型を示すため、遺伝子型・表現型相関を参考にしつつも臨床的な個別評価が欠かせません。

3. 主な症状と全身合併症
FAPは大腸の病気と思われがちですが、実際には全身の多臓器に多彩な腫瘍素因をもたらす症候群です。大腸のサーベイランスだけでは患者さんの命を守りきれません。
大腸の症状
古典型FAPでは平均16歳(範囲7〜36歳)から大腸内に腺腫が発生し始め、35歳までに95%の患者さんに多数のポリープが認められます。初期は無症状ですが、進行すると血便・便通異常・腹痛・貧血が出現します。未治療の場合、大腸がん発症の平均年齢は39歳(範囲34〜43歳)に達します。
特に注目すべき臨床的事実として、予防的結腸切除術を施行される前のFAP患者の90%において直腸がんが先行して認められることが報告されており、直腸の管理がFAPの予後を大きく左右します。
FAPで増加する大腸外悪性腫瘍:生涯リスク
🔴 大腸がん
- 生涯リスク:ほぼ100%(未治療)
- 結腸がんHR:2.16〜2.72
- 直腸がんHR:9.12〜12.66
🟡 十二指腸・ファーター乳頭がん
- 生涯リスク:最大18%
- 一般人口は1%未満
- 大腸切除後の主要死因の一つ
🔵 胃がん
- 生涯リスク:最大7%
- GAPPS型ではさらに高リスク
- 一般人口0.8%
🟣 甲状腺乳頭がん
- 生涯リスク:1.2〜12%
- 女性に好発(特殊型CMTC)
- 10代後半から年1回の検診を推奨
🟢 肝芽腫
- 生涯リスク:0.4〜2.5%
- 5歳未満の男児に好発
- AFP・腹部超音波でモニタリング
🟠 膵臓がん・脳腫瘍
- 膵がんHR:7.66
- 中枢神経系腫瘍:約1%
- 大腸外合併がん全体:10〜15%
良性病変も見逃せない
FAPでは悪性腫瘍以外にも、デスモイド腫瘍(線維性腫瘍。手術侵襲が引き金)、先天性網膜色素上皮肥大(CHRPE)、骨腫(特に下顎・頭蓋)、歯科異常(過剰歯・埋伏歯)、副腎腫瘍など、多彩な良性病変が随伴します。これらの一部はFAPの早期診断のサインとなることもあるため、関連する他科の医師との情報共有が重要です。
🔍 関連記事:FAPに合併する遺伝性デスモイド腫瘍や肝芽腫、進行した場合の大腸がんについては各専門ページをご覧ください。
4. FAPのサブタイプ・関連症候群と鑑別診断
FAPと類縁の病態には多くのサブタイプ・関連症候群が存在します。それぞれを正確に区別することは、その後のサーベイランス計画と治療方針を決めるうえで不可欠です。
減弱型FAP(AFAP)との違い
特徴:大腸ポリープが100個未満で右側結腸寄り。発症年齢は遅い。
注意:「軽症」と思われがちですが、生涯発がんリスクは依然として約70%。AFAPでも厳重なサーベイランスが必要です。
MUTYH関連ポリポーシス(MAP)との鑑別
特徴:MUTYH遺伝子の両アレル変異による類似疾患。大腸に多発ポリープ(通常15〜100個)が生じ、表現型はAFAPに酷似します。
鑑別の鍵:遺伝形式が常染色体潜性(劣性)。親から子へ直接は伝わりにくく、両親が保因者でも無症状で、きょうだいに同じ病気が出る(水平発症)パターンをとります。家族歴の出方がFAPと根本的に異なります。
ガードナー症候群
特徴:FAPに加えて、骨腫・デスモイド腫瘍・類表皮嚢腫・歯科異常などの大腸外所見が顕著なサブタイプ。
鑑別の鍵:同じAPC変異でも、特定領域(特に3’側)の変異で大腸外症状が前景化します。
ターコット症候群
特徴:消化管ポリポーシスと中枢神経系腫瘍(髄芽腫など)を合併する稀少疾患。
鑑別の鍵:APC変異型(タイプ2)とリンチ症候群型(タイプ1)が存在し、原因遺伝子が異なります。
GAPPS(胃近位部ポリポーシス)
特徴:APCプロモーター1Bの点変異により、胃の近位部にのみ絨毯状のポリポーシスが発生。大腸・十二指腸はほぼ無病変。
鑑別の鍵:胃腺がんリスクが極めて高い。標準FAPの大腸サーベイランス基準は適用されません。
遺伝性デスモイド腫瘍
特徴:大腸ポリポーシスを伴わず、デスモイド腫瘍のみが家族性に発生する病型。
鑑別の鍵:APC遺伝子の特定領域の変異が原因。FAPと同じ遺伝子でも臨床像が大きく異なります。
5. 診断と遺伝子検査の進め方
FAPの診断は、臨床的所見と遺伝学的検査の両面から行われます。早期診断と正確な変異同定は、患者さん本人だけでなく、血縁者の発症前診断・サーベイランス計画にも直結します。
FAPを疑うべき臨床的所見
💡 こういった所見があればFAPを強く疑います
- ➤大腸内視鏡で100個以上の腺腫性ポリープを認める
- ➤40歳未満で複数の腺腫性ポリープが見つかった
- ➤FAPの第一度近親者がいる
- ➤50歳未満で大腸がんを発症した
- ➤デスモイド腫瘍・骨腫・CHRPE・甲状腺乳頭がん・肝芽腫など、FAPを示唆する大腸外病変
診断アプローチの国際的差異:欧米と日本(JSCCR)
FAPの診断方針については、欧米の主要ガイドライン(NCCN・ACG・BSG/ESGE)と、日本の大腸癌研究会(JSCCR)ガイドラインとの間に、明確な考え方の違いがあります。日本で診療を受ける方が知っておくと役立つポイントです。
🌐 欧米アプローチ(NCCN・ACG・BSG)
ゲノムを起点とする戦略:少数のポリープでも早期に遺伝学的背景を解明します。BSG/ACPGBIガイドラインでは、60歳未満で生涯10個以上の累積結腸腺腫を有する患者に対し生殖細胞系列遺伝子検査を強く推奨。MUTYH関連ポリポーシス(MAP)やPPAPなどの鑑別除外を優先します。
🇯🇵 日本アプローチ(JSCCR)
臨床所見を第一とするアプローチ:「典型的なFAP」は100個以上の大腸腺腫という臨床所見のみで確定診断が可能で、ルーチンの遺伝子検査は必須としません。遺伝子検査(APC検査含む)は、AFAPとMAPの臨床的鑑別が困難な場合や、患者さんが確定診断を希望する場合に提案されます。
💡 用語解説:MUTYH関連ポリポーシス(MAP)との鑑別が重要な理由
FAPと臨床像が似ているMAPは、MUTYH遺伝子の両アレル変異によって発症する常染色体潜性(劣性)遺伝疾患です。腺腫数が少なく(通常15〜100個)、親から子への直接伝達は起こりにくく、きょうだい間で発症リスクが上がります。腺腫数が10〜99個でAFAPと一致する範囲の場合、MAPとの鑑別を目的に遺伝子検査が必要です。遺伝形式が違えば、ご家族のどなたを検査すべきかという方針も根本的に変わります。
遺伝学的検査:APC遺伝子解析
💡 用語解説:生殖細胞系列変異(せいしょくさいぼうけいれつへんい)
受精卵の段階から全身のすべての細胞に存在する変異のことで、子どもへ受け継がれる可能性があります。これに対し、生まれた後にがん組織だけで生じる変異は「体細胞変異」と呼ばれ、子どもには遺伝しません。FAPの診断には血液や唾液から採取したDNAを用いた生殖細胞系列の変異検査が行われます。
遺伝学的検査では、まずAPC遺伝子全体のシーケンス解析と、欠失・重複を検出するためのMLPA法などが組み合わせて行われます。APCで変異が同定されない場合には、MUTYH遺伝子の解析や、より広範な遺伝性腫瘍包括的パネル検査、あるいは全エクソーム解析(WES)へとステップアップしていきます。
家族内で病的バリアントが既に同定されている場合、血縁者の発症前診断は家族内既知変異の有無のみを確認するシンプルな検査で済むため、コストと時間を大幅に節約できます。家系内で誰か一人を最初に検査するときの「インデックスケース解析」が、その後の家族全員の管理計画の出発点になります。
未成年への遺伝子検査の特殊性
FAPは10代から大腸内視鏡サーベイランスを開始する必要があるため、子どもの段階での遺伝子検査が医学的に正当化される稀な遺伝性腫瘍の一つです。検査を受けた未成年の子どもの心理的サポートや、結果開示のタイミング・方法については、専門の遺伝カウンセラー・臨床遺伝専門医による事前の十分な準備と継続的な支援が不可欠です。
6. 生涯にわたるサーベイランス計画
未治療のFAPはほぼ100%の確率で致死的な大腸がんへ進行するため、無症候期からの厳格なサーベイランスが患者さんの予後を決定する唯一の手段です。米国NCCN・欧州ESGE・英国BSGなどのガイドラインに基づき、表現型ごとに細やかな計画が立てられます。
大腸サーベイランス:開始年齢と頻度
| 表現型 | 開始年齢 | 頻度 | 術後フォロー |
|---|---|---|---|
| 古典型FAP | 10〜15歳 | 1〜2年ごと | 術後6〜12ヶ月から、6〜12ヶ月ごと |
| 減弱型FAP(米国) | 18〜20歳 | 1〜2年ごと | 同上 |
| 減弱型FAP(欧州) | 12〜14歳 | 1〜3年ごと | 術後から1〜3年ごと |
上部消化管サーベイランス:Spigelman分類による階層化
大腸を予防的に切除した後のFAP患者さんでは、十二指腸がんが主要な死因として浮上します。胃カメラ・十二指腸内視鏡検査は遅くとも20〜25歳までには開始し、十二指腸ポリポーシスの重症度評価には世界標準のSpigelman(スピーゲルマン)分類が用いられます。
💡 用語解説:Spigelman分類とは
十二指腸ポリポーシスの重症度を客観的にスコア化するためのシステムです。①ポリープの数 ②サイズ ③組織型 ④異型度の4項目それぞれに1〜3点を付け、合計点数でStage 0〜IVの5段階に分類します。次回の内視鏡検査の間隔や、内視鏡治療・外科治療の必要性を判断するための国際的な共通言語として機能しています。
| 評価項目 | 1点 | 2点 | 3点 |
|---|---|---|---|
| ポリープの数 | 1〜4個 | 5〜20個 | 20個超 |
| サイズ | 1〜4mm | 5〜10mm | 10mm超 |
| 組織型 | 管状 | 管状絨毛 | 絨毛 |
| 異型度 | 軽度 | 中等度 | 高度 |
Spigelman分類に基づく管理プロトコル
Stage 0〜I(0〜4点)
低リスク群。3〜5年ごとの内視鏡検査で十分。10年以内のがん化は極めて稀。積極的介入は原則不要。
Stage II(5〜6点)
中等度リスク群。2〜3年ごとの内視鏡検査。化学予防の併用が考慮され始めるステージ。
Stage III(7〜8点)
高リスク群。6〜12ヶ月ごとの厳密なサーベイランス。1cm超の病変や高度異形成には内視鏡的粘膜切除を検討。
Stage IV(9〜12点)
極高リスク群。10年以内の浸潤がん進行リスクは36%。膵頭十二指腸切除術などの外科介入を検討。
十二指腸ポリポーシスの重症度は加齢とともに不可逆的に進行する性質があり、Stage IVの十二指腸ポリポーシスを発症する累積リスクは60歳までに最大43%、70歳までに50%に達すると推定されています。なお、Spigelman分類はファーター乳頭部病変を含まないため、内視鏡医は側視鏡と直視鏡を併用してファーター乳頭部を含む十二指腸全体を詳細に評価する必要があります。
大腸外病変のスクリーニング
- 🦋 甲状腺:10代後半から年1回の触診と超音波検査
- 💪 デスモイド腫瘍:年1回の腹部触診。家族歴あり・術後はCT/MRIで定期的画像評価
- 👶 肝芽腫:5歳未満の男児で3〜6ヶ月ごとの肝臓超音波検査とAFP測定
- 👁 CHRPE:FAPを示唆する眼科所見。眼底検査で確認可能
甲状腺がん(CMTC)と肝芽腫:見落としやすい大腸外がんの具体的監視法
大腸外のがんのうち、FAP特有のパターンを示すのが甲状腺乳頭がんの特殊型と、乳幼児期の肝芽腫です。いずれも好発年齢が大きく異なるため、それぞれに合わせた監視のタイミングが重要になります。
💡 用語解説:クリブリフォルム・モリュラー型甲状腺がん(CMTC)
FAP患者に高頻度に見られる甲状腺乳頭がんの特殊な組織型で、WHO第5版分類ではCMTC(旧称CMV-PTC)と呼ばれます。通常型の甲状腺乳頭がんが平均45歳・女性対男性3対1で発症するのに対し、CMTCは平均23〜28歳という非常に若い年齢で、女性対男性31対1と圧倒的に女性に好発します。CMTC症例の40〜53%にFAPが合併すると報告されており、若年女性のCMTCはFAP発見の手がかりにもなります。リンパ節転移は少なく、多くは多中心性に発生し、長期予後は良好です。
🦋 甲状腺がん(CMTC)の監視
開始年齢:16歳(10代後半)から。特に女性は重点的に。
方法・頻度:甲状腺超音波検査を2〜5年ごと(施設により年1回)。
治療:甲状腺全摘が基本。予防的な頸部リンパ節郭清は通常不要とされます。
👶 肝芽腫の監視
好発時期:ほとんどが生後30ヶ月(2歳半)以前に発症。
方法・頻度:出生時〜診断時から5〜7歳まで、3〜6ヶ月ごとに血清AFP測定+腹部超音波。
ポイント:患者の95%以上でAFPが上昇。早期発見は外科切除の成功率を高め、強い化学療法を回避できます。
7. 治療:予防的手術と化学予防
FAP患者さんが大腸がんによる死を確実に回避するための唯一の根治的治療は予防的大腸切除術です。一方、20歳以前に大腸がんを発症することは極めて稀なため、手術のタイミング選択においては「がん死回避」と「思春期の生活の質・生殖機能・成長への影響を最小限にする」という相反する要請のバランスが求められます。
外科治療の二大標準術式:IPAA vs IRA
🔴 IPAA:回腸嚢肛門吻合術を伴う大腸全摘術
結腸・直腸の粘膜をすべて切除し、回腸でJ型などのパウチを作って肛門管に吻合する根治的術式。
利点:直腸がんリスクをほぼ排除できる。
欠点:排便回数増加・夜間排便・便失禁が頻発。骨盤内深部の操作で性機能・妊孕性への影響リスク。パウチ不全率5〜15%。
🔵 IRA:回腸直腸吻合術を伴う結腸全摘術
結腸のみを切除し、残した直腸に回腸を直接吻合する機能温存術式。
利点:排便機能・性機能・妊孕性が維持されやすい。術後QoLが優れる。
欠点:残存直腸からの発がんリスク。3〜6ヶ月ごとの厳格な直腸内視鏡が生涯必要。約1/3が後に補完的直腸切除へ。
直腸に高度のポリポーシスがすでに形成されている場合は最初からIPAAを選択しますが、直腸の病変が比較的軽度で患者さんが厳格なフォローアッププログラムを遵守できる場合には、QoLと機能温存の観点からIRAを第一選択とすることが、現代の個別化医療として推奨されています。なお、APC遺伝子検査でデスモイド腫瘍の高リスク変異(コドン1395〜1493など)が判明している場合は、手術侵襲がデスモイド発症の引き金になりうるため、侵襲の少ない術式の選択や手術時期の検討がより慎重に行われます。
GAPPSにおける新しい胃切除戦略
GAPPSでは胃の近位部に病変が限定されるという特徴を活かし、従来の「胃全摘」に代わって「二重トラクト再建を伴う近位胃切除術」が新たな選択肢として施行されています。これにより術後のQoL(体重減少・ダンピング症候群・栄養吸収障害の軽減)を担保しながら、残存する遠位胃や十二指腸への内視鏡的サーベイランスを継続できる解剖学的構造が維持されます。
化学予防(Chemoprevention)の現状と限界
予防的手術に代わる、あるいは手術までの期間を延長するための薬物療法(化学予防)は、FAP管理における長年の課題です。
スリンダク(NSAIDs)
既存の腺腫の数を約50%、サイズを約65%減少させる効果が報告されています。ただし投薬中止で再発するため根治薬ではなく、新たな腺腫の発生そのものを予防する効果は確認されていません。IRA術後の直腸ポリープ管理に使用されます。
セレコキシブ(COX-2阻害薬)
かつてポリープ数・サイズの減少が報告されましたが、FAPに対する適応はその後取り下げられ、現在は標準的な位置づけではありません。化学予防はあくまでサーベイランスの補助です。
エフロルニチン+スリンダク
第3相FAP-310試験で評価されましたが、併用群と単独療法群で疾患進行の有意差は認められませんでした(NEJM 2020)。FAP化学予防の難しさを示す象徴的な結果となりました。
mTOR阻害薬(ラパマイシン/eRapa)
第2相試験で75%の患者が「非進行」と判定。ポリープ負担の中央値で17%減少。次世代の化学予防候補として注目されています。
デスモイド腫瘍の最新治療:ニロガセスタット(2023年FDA承認)
大腸がんを外科的に予防できるようになった現代のFAPでは、デスモイド腫瘍が十二指腸がんと並ぶ主要な死因として浮上しています。デスモイド腫瘍は転移しない良性の線維性腫瘍ですが、局所浸潤性が極めて強く、腸管・血管・神経を巻き込んで激しい疼痛や臓器障害を引き起こします。従来は外科切除が第一選択でしたが、手術そのものが新たなデスモイドの引き金になるというジレンマがありました。
2023年11月、米国FDAは進行性デスモイド腫瘍の成人患者に対する世界初の全身療法薬として、経口γセクレターゼ阻害薬ニロガセスタット(商品名OGSIVEO)を承認しました。これはFAP関連デスモイド腫瘍の治療における大きな転換点です。
DeFi試験:デスモイド腫瘍に対するニロガセスタットの客観的奏効率(ORR)
ニロガセスタット群
(n=71)
プラセボ群
(n=71)
無増悪生存期間(PFS):ニロガセスタット群は中央値未到達。プラセボ群の中央値15.1ヶ月と比較して、病勢進行・死亡リスクを71%減少(ハザード比0.29、95%CI 0.15-0.55、p<0.001)。
💡 用語解説:γセクレターゼ阻害薬(ガンマセクレターゼそがいやく)
γセクレターゼとは、細胞内でNotchなどのタンパク質を切断・活性化する酵素複合体です。デスモイド腫瘍ではNotch経路が過剰活性化しており、これを阻害することで腫瘍細胞の増殖・生存を抑制します。ニロガセスタットは150mgを1日2回経口投与する分子標的薬で、根拠となったDeFi第3相試験(NCT03785964、切除不能な進行性デスモイド腫瘍患者142名)では疾患進行リスクを71%低下させました。下痢・卵巣毒性・発疹・悪心・疲労などの副作用が報告されていますが、忍容性は高いと判断されています。日本国内での承認状況は変動するため、使用にあたっては最新情報を担当医にご確認ください。
8. 遺伝カウンセリングと家族計画
FAPの確定診断後、患者さん本人だけでなくご家族全員にとって、遺伝カウンセリングは重要なステップとなります。検査前から検査後の意思決定支援、そして次世代の家族計画まで、長期的に伴走する仕組みです。
- ➤遺伝形式と再発リスクの説明:常染色体顕性遺伝のため、患者さんの子どもへの遺伝確率は理論上50%。約25〜30%の症例ではde novo(新生)変異で、両親に変異がない場合もあります。
- ➤血縁者の発症前診断:家族内で既知の病的変異がある場合、血縁者は単純な変異確認検査でリスクを評価できます。陰性なら一般人口と同等のリスクに、陽性なら計画的なサーベイランスへ。
- ➤家族計画と出生前診断:次世代へ変異を伝えたくない方に対しては、単一遺伝子疾患の出生前診断や着床前遺伝学的検査(PGT-M)が選択肢となります。
- ➤未成年への結果開示:10代から大腸サーベイランスが必要なため、成熟度に応じた段階的な情報共有と心理的サポートが不可欠です。
- ➤長期的フォローアップ:がんの予防だけでなく、就学・就労・保険加入など社会生活上の課題にも継続的に伴走します。
🔍 関連記事:遺伝性疾患のキャリア(保因者)スクリーニングについてはキャリアスクリーニングとはと米国人類遺伝学会の推奨内容もあわせてご覧ください。
9. よくある誤解
誤解①「家族にFAPの人がいないから自分は大丈夫」
FAP症例の約25〜30%はde novo(新生)変異によるもので、両親に変異がない場合があります。ご自身に多発ポリープや若年性大腸がんがあれば、家族歴がなくても遺伝子検査の意義は十分にあります。
誤解②「大腸を全部取れば、もう何も心配しなくていい」
大腸切除後も、十二指腸がん・甲状腺がん・デスモイド腫瘍・パウチ内の新規腺腫など多彩な合併症リスクが残ります。むしろ術後こそ全身サーベイランスが本格化します。
誤解③「減弱型(AFAP)は軽症だから手術はいらない」
AFAPでも生涯発がんリスクは約70%に達します。発症が遅いだけで、サーベイランスや必要に応じた予防的手術の重要性は古典型FAPと変わりません。
誤解④「化学予防の薬を飲めば手術は不要になる」
スリンダクやセレコキシブなどの化学予防はポリープを減らす効果はあっても根治には至らず、薬を中止すれば再発します。現時点で予防的大腸切除を完全に代替できる薬剤は存在しません。
10. 臨床遺伝専門医からのメッセージ
FAPは、遺伝子診断技術の進歩と多診療科の連携によって「予防可能な遺伝性がん」の代表例となりました。発症前から正しい知識を持ち、計画的にサーベイランスを行えば、大腸がんによる死亡はほぼ確実に回避できます。
同時に、FAPは家系全体の健康管理が問われる疾患でもあります。一人の確定診断が、その方の親・きょうだい・子どもたちの人生を変える情報をもたらします。だからこそ、遺伝カウンセリングと臨床遺伝専門医による継続的な伴走が欠かせません。ミネルバクリニックでは、FAPおよび関連症候群の遺伝子検査・遺伝カウンセリング・他施設との連携医療をワンストップで提供しています。少しでも気になることがあれば、どうぞお気軽にご相談ください。
よくある質問(FAQ)
🏥 FAP・遺伝性大腸がんのご相談はミネルバクリニックへ
家族性大腸腺腫症(FAP)をはじめとする遺伝性腫瘍に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] Familial Adenomatous Polyposis. StatPearls (NCBI Bookshelf). [NCBI Bookshelf NBK538233]
- [2] APC-Associated Polyposis Conditions. GeneReviews® (NCBI Bookshelf). [NCBI Bookshelf NBK1345]
- [3] Familial adenomatous polyposis. MedlinePlus Genetics. [MedlinePlus]
- [4] Cancer risks in familial adenomatous polyposis. American College of Gastroenterology (EBGI). [ACG EBGI]
- [5] Mechanism of APC truncation involved in colorectal cancer tumorigenesis (Review). Oncology Letters. 2024. [Spandidos Publications]
- [6] The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. The Application of Clinical Genetics. [PMC4404874]
- [7] Guidelines for Familial Adenomatous Polyposis (FAP): challenges in defining clinical management for a rare disease. Familial Cancer. 2025. [PMC11976741]
- [8] Surveillance of Duodenal Adenomas in Familial Adenomatous Polyposis Reveals High Cumulative Risk of Advanced Disease. Journal of Clinical Oncology. [JCO]
- [9] Surgical management of the colorectum in FAP: tailored approaches for optimal outcomes. PMC. [PMC12413404]
- [10] Familial adenomatous polyposis: non-surgical management of large bowel disease: endoscopic and chemoprevention strategies. PMC. 2025. [PMC12127233]
- [11] Eflornithine plus Sulindac for Prevention of Progression in Familial Adenomatous Polyposis. NEJM. 2020. [PubMed 32905675]
- [12] Consideration of a Novel Surgical Approach in the Management of Gastric Adenocarcinoma and Proximal Polyposis Syndrome. JCO Precision Oncology. [ASCO]
- [13] FDA approves nirogacestat for desmoid tumors. U.S. Food and Drug Administration. 2023. [FDA]
- [14] Cribriform-morular variant of papillary thyroid carcinoma and its association with familial adenomatous polyposis. PMC. [PMC6650210]
- [15] Oncogenic mutations in adenomatous polyposis coli (Apc) activate mTORC1 in mice and zebrafish. Disease Models & Mechanisms. [DMM]






