目次
副腎白質ジストロフィーとは
副腎白質ジストロフィー(Adrenoleukodystrophy, ALD)とは
副腎白質ジストロフィー(Adrenoleukodystrophy, ALD) は、X連鎖性遺伝 によって引き起こされる神経変性疾患であり、主に中枢神経系(脳の白質)と副腎 に影響を与えます。
この疾患は、極長鎖脂肪酸(VLCFA) を分解する酵素が正常に機能しないことにより、脂肪酸が細胞内に蓄積し、神経細胞や副腎皮質に障害を引き起こします。
副腎白質ジストロフィーの特徴
遺伝的背景
X染色体にある ABCD1遺伝子 の変異 が原因で発症します。男性に多く、女性はキャリア(保因者)として軽度の症状を示すことがあります。
主な病型
ALDには以下の3つの主要な病型があります。
- 小児大脳型ALD:主に 3〜10歳の男児 に発症し、神経症状が急速に進行する最も重篤なタイプ。
- 副腎脊髄ニューロパチー(AMN):成人男性に多く、運動障害や感覚異常が徐々に進行。
- 副腎不全型:副腎皮質ホルモンの分泌障害が主な症状となるタイプ。
副腎白質ジストロフィーの影響
この疾患は 進行性 であり、特に 小児大脳型ALD では、治療を行わなければ数年以内に重度の障害や死亡 に至る可能性があります。
そのため、早期発見と適切な治療 が極めて重要です。
副腎白質ジストロフィーの診断と治療
診断方法
診断方法 には、以下のような検査が用いられます。
- 血液検査:極長鎖脂肪酸(VLCFA)の測定
- 遺伝子検査:ABCD1遺伝子の変異を特定
- MRI検査:脳の白質の評価
治療法
ALDの治療法として、以下の方法が挙げられます。
- 造血幹細胞移植:小児大脳型ALDの進行抑制に有効
- 副腎皮質ホルモン補充療法:副腎不全の管理
- ロレンツォオイル療法:食事療法の一環
副腎白質ジストロフィーの早期発見の重要性
近年、新生児スクリーニング を導入する国が増えており、早期発見による適切な治療介入が可能になりつつあります。
ALDの進行を抑えるためには、定期的な健康診断 と 家族歴の把握 が重要です。
副腎白質ジストロフィーの原因
副腎白質ジストロフィー(Adrenoleukodystrophy, ALD) は、X染色体にある ABCD1遺伝子 の変異 によって引き起こされる遺伝性疾患です。
ABCD1遺伝子は、細胞内のペルオキシソームという小器官に存在するALDタンパク質(ALDP)をコードしており、このタンパク質は極長鎖脂肪酸(VLCFA)を代謝する役割を持っています。
ABCD1遺伝子が変異すると、ALDPの機能が失われ、VLCFAが細胞内に蓄積します。
この蓄積が神経細胞や副腎皮質に障害を引き起こし、神経変性や副腎不全をもたらします。
副腎白質ジストロフィーの遺伝的背景
X連鎖性遺伝の仕組み
副腎白質ジストロフィーはX連鎖性遺伝疾患であり、主に男性に発症します。その理由は、以下の通りです。
- 男性(XY):X染色体を1本しか持たないため、ABCD1遺伝子に変異があると症状が発症する。
- 女性(XX):2本のX染色体を持っているため、片方のX染色体が正常であれば軽症または無症状となる(キャリア)。
ただし、キャリア女性 でも加齢とともに軽度の神経症状が現れることがあります。
ABCD1遺伝子の変異パターン
現在、ABCD1遺伝子の変異は2,000種類以上が報告されており、変異の種類によってALDの病型や重症度に違いが生じる可能性があります。
しかし、同じ遺伝子変異を持っていても発症年齢や症状の進行度には個人差があるため、遺伝的な予測が難しいのが特徴です。
副腎白質ジストロフィーの発症メカニズム
- ABCD1遺伝子の変異 → ALDPの機能が失われる
- 極長鎖脂肪酸(VLCFA)の蓄積 → 細胞内での毒性が増加
- 中枢神経系の白質(ミエリン)の破壊 → 神経伝達の障害
- 副腎皮質の機能不全 → ホルモン不足による体調不良
このプロセスが進行すると、運動機能の低下、認知機能の障害、副腎不全などの症状が現れます。
遺伝カウンセリングの重要性
副腎白質ジストロフィーは遺伝性疾患のため、家族内で発症リスクを把握することが大切です。
遺伝カウンセリングを受けることで、以下の点を明確にできます。
- 家族内のリスク評価(兄弟や親族の発症リスクの判定)
- キャリア女性の検査(女性が保因者かどうかを判定)
- 出生前診断の選択肢(妊娠中に胎児の遺伝子を調べる)
特に、ALDの新生児スクリーニングが導入されつつあり、早期診断と適切な治療の重要性が高まっています。
副腎白質ジストロフィーの症状
小児大脳型ALDの特徴
小児大脳型副腎白質ジストロフィー(Childhood Cerebral Adrenoleukodystrophy, cALD) は、副腎白質ジストロフィー(ALD)の中で最も重篤な病型 であり、主に3〜10歳の男児 に発症します。
この病型は、急速に進行する神経症状を特徴とし、早期に診断し適切な治療を行わなければ、数年以内に重度の障害や死に至る可能性があります。
小児大脳型ALDの主な症状
小児大脳型ALDの初期症状は非特異的であり、注意欠陥や学習障害として現れることが多いため、診断が遅れることがあります。
進行すると以下のような症状が現れます。
- 学習障害や注意力の低下:学校での成績低下や集中力の欠如
- 行動変化:攻撃的な行動、衝動性の増加
- 視力や聴力の低下:徐々に進行する感覚障害
- 運動障害:歩行困難、協調運動の障害
- 言語能力の低下:会話が困難になる
- けいれん発作:てんかん発作の発症
症状が進行すると、認知機能の著しい低下 や 運動麻痺 を引き起こし、最終的には寝たきりの状態になることもあります。
小児大脳型ALDの進行と予後
小児大脳型ALDは急速に進行するため、診断が遅れると治療の選択肢が限られてしまいます。
一般的には、発症後2〜5年以内に重度の障害を伴い、最終的には生命を脅かす状態に至ることが多いとされています。
ただし、早期診断と適切な治療を行うことで、病状の進行を抑えることが可能です。
小児大脳型ALDの診断方法
小児大脳型ALDの診断には、以下の検査が用いられます。
- 血液検査:極長鎖脂肪酸(VLCFA)の蓄積を測定
- 遺伝子検査:ABCD1遺伝子の変異を確認
- MRI検査:脳の白質の病変を確認
MRI検査では、初期段階から脳の後頭葉や側頭葉の白質に病変が見られ、これが診断の決め手となります。
小児大脳型ALDの治療法
小児大脳型ALDに対する治療法には以下のものがあります。
- 造血幹細胞移植(HSCT):病気の進行を抑える最も有効な治療法
- 副腎皮質ホルモン補充療法:副腎機能不全がある場合に必要
- ロレンツォオイル療法:食事療法として用いられるが効果は限定的
造血幹細胞移植は、病気の進行が軽度の段階で行うことで、神経症状の悪化を防ぐ効果が期待できます。
小児大脳型ALDの早期発見の重要性
近年、多くの国で新生児スクリーニングが導入され、早期診断と治療が可能になりつつあります。
小児大脳型ALDの早期発見は、適切な時期に造血幹細胞移植を行うために不可欠です。
家族にALDの既往がある場合は、遺伝カウンセリングを受けることで、発症リスクを把握し、適切な対策を講じることができます。
副腎脊髄ニューロパチー(AMN)の症状
副腎脊髄ニューロパチー(Adrenomyeloneuropathy, AMN) は、副腎白質ジストロフィー(ALD)の成人型として知られ、主に成人男性に発症する病型です。
ALDの中でも最も一般的なタイプであり、30〜40代で発症することが多く、進行性の神経障害を引き起こします。
副腎脊髄ニューロパチー(AMN)の主な症状
AMNの症状は、神経系の進行性変性と副腎機能不全を特徴とし、以下のような症状が現れます。
- 歩行障害:両脚の筋力低下やこわばり(痙性麻痺)により、歩行が困難になる
- バランス障害:転倒しやすくなり、階段の昇降が難しくなる
- 感覚異常:下肢のしびれや痛み、感覚低下
- 膀胱・直腸障害:頻尿や尿失禁、便秘などの排泄障害
- 副腎機能不全:コルチゾール分泌の低下による倦怠感、低血圧、色素沈着
初期の症状は軽度であることが多いですが、時間の経過とともに進行し、車椅子が必要になるケースもあります。
副腎脊髄ニューロパチー(AMN)の進行と予後
AMNは慢性的に進行するため、発症後数十年にわたり徐々に悪化します。
一部の患者では、小児大脳型ALDのような脳白質病変(炎症性ALD)が発生し、重度の神経症状が急速に進行することがあります。
また、副腎機能不全が未治療のまま放置されると、命に関わる危険性があるため、定期的なホルモン検査が推奨されます。
副腎脊髄ニューロパチー(AMN)の診断方法
AMNの診断には、以下の検査が用いられます。
- 血液検査:極長鎖脂肪酸(VLCFA)の測定
- 遺伝子検査:ABCD1遺伝子の変異を確認
- MRI検査:脊髄の萎縮や脳白質病変の有無を確認
- 神経伝導検査:末梢神経の障害を評価
- 内分泌検査:副腎機能不全の有無を確認
これらの検査を組み合わせることで、AMNの正確な診断が可能になります。
副腎脊髄ニューロパチー(AMN)の治療法
現在、AMNを完治させる治療法はありませんが、症状の進行を遅らせる治療が行われます。
- 副腎皮質ホルモン補充療法:副腎機能不全がある場合、コルチゾール補充を行う
- 理学療法:リハビリを通じて筋力低下を抑え、歩行能力を維持
- 対症療法:痙性麻痺や疼痛を軽減するための薬物療法
- 造血幹細胞移植:脳白質病変が進行する場合に適応されることがある
- 遺伝子治療:現在、臨床試験が進行中
これらの治療を適切に行うことで、生活の質(QOL)の維持が可能となります。
副腎脊髄ニューロパチー(AMN)の早期発見の重要性
AMNは早期診断と適切な管理が重要な疾患です。
進行を抑えるために、定期的な神経検査やホルモン検査を受けることが推奨されます。
また、ALDの家族歴がある場合は、遺伝カウンセリングを受けることで、発症リスクの評価や早期対策が可能になります。
副腎機能不全の兆候
副腎機能不全(Adrenal Insufficiency) は、副腎白質ジストロフィー(ALD)において高頻度で発生する合併症であり、副腎皮質ホルモンの不足によりさまざまな症状を引き起こします。
ALD患者の約90%が副腎機能不全を発症するとされており、特に小児大脳型ALDや副腎脊髄ニューロパチー(AMN)の患者に多く見られます。
副腎機能不全の主な症状
副腎機能不全の症状は、コルチゾールやアルドステロンなどの副腎皮質ホルモンの欠乏によって引き起こされます。以下のような兆候が現れた場合、早急な医療対応が必要です。
- 慢性的な疲労感:倦怠感が強く、日常生活に支障をきたす
- 低血圧:立ちくらみやめまいが頻繁に起こる
- 食欲不振と体重減少:栄養不足につながる
- 皮膚の色素沈着:手のひらのしわや歯茎などが黒ずむ
- 低血糖:倦怠感や意識障害を引き起こす
- 塩分の欲求増加:ナトリウム不足によるもの
- 吐き気や腹痛:消化器系の不調
- 精神的な不安やうつ症状:ホルモンバランスの乱れによる影響
これらの症状は徐々に進行することが多いため、早期発見と適切な治療が重要です。
副腎クリーゼ(アジソンクリーゼ)の危険性
副腎機能不全が進行すると、副腎クリーゼ(アジソンクリーゼ)と呼ばれる生命を脅かす急性症状が発生することがあります。
- 突然の激しい腹痛や嘔吐
- 急激な血圧低下とショック症状
- 意識障害
- 発熱や極度の脱力感
副腎クリーゼは緊急の治療が必要であり、発症した場合はすぐに医療機関を受診することが重要です。
副腎機能不全の診断方法
副腎機能不全の診断には、以下の検査が行われます。
- 血液検査:コルチゾールやACTH(副腎皮質刺激ホルモン)の測定
- ACTH刺激試験:副腎が適切にホルモンを分泌できるかを評価
- 電解質検査:ナトリウムやカリウムのバランスを確認
- 副腎CTやMRI:副腎の萎縮や異常を確認
副腎機能不全の治療法
副腎機能不全は、ホルモン補充療法によって管理されます。
- ヒドロコルチゾン(コルチゾール補充):ストレス時には増量が必要
- フルドロコルチゾン(アルドステロン補充):ナトリウムとカリウムのバランスを維持
- 緊急時のステロイド注射:副腎クリーゼの発症時に使用
これらの治療を適切に行うことで、生活の質(QOL)を維持しながら健康を管理することが可能です。
副腎機能不全の早期発見の重要性
副腎機能不全は、早期に診断し適切な治療を受けることで、深刻な合併症を防ぐことができます。
ALDの患者や家族は、定期的な血液検査を受け、副腎機能の低下を早期に発見することが重要です。
また、ストレスや感染症などが副腎クリーゼを引き起こすリスク要因となるため、注意が必要です。
家族歴がある場合や、ALDと診断された場合は、専門医によるフォローアップを受けることを推奨します。
副腎白質ジストロフィーの診断方法
血液検査による極長鎖脂肪酸(VLCFA)の測定
極長鎖脂肪酸(Very Long Chain Fatty Acids, VLCFA) は、副腎白質ジストロフィー(ALD)の診断において最も重要なバイオマーカーの一つです。
ALDの患者では、細胞内のペルオキシソームが正常に機能しないため、VLCFAが分解されずに蓄積します。
そのため、血液検査による極長鎖脂肪酸の測定は、ALDの早期診断に不可欠な検査となっています。
極長鎖脂肪酸(VLCFA)とは?
VLCFAは、炭素鎖が22炭素以上の長さを持つ脂肪酸のことを指します。
これらの脂肪酸は、通常ペルオキシソーム内で分解されますが、ALD患者では代謝が阻害されるため、血液中に異常に高い濃度で蓄積します。
- C24:0(リグノセリン酸)
- C26:0(セロチン酸):ALDの診断において最も重要な指標
- C22:0(ベヘン酸)
これらの脂肪酸の比率を測定することで、ALDの可能性を高精度で評価することができます。
血液検査によるVLCFA測定の目的
ALDの診断では、血漿または赤血球膜中のVLCFAレベルを測定することで、以下の点を明らかにできます。
- ALDのスクリーニング:症状が現れる前の早期診断
- 遺伝子検査の補助:ABCD1遺伝子の変異があるかを確認
- 治療の経過観察:VLCFAの変動をチェックし、病状の進行を評価
VLCFA測定の基準値と異常値
血液中のVLCFA濃度は、特定の基準値を超えると異常とみなされ、ALDの可能性が高くなります。
| 脂肪酸の種類 | 正常値 | ALD患者の異常値 |
|---|---|---|
| C24:0/C22:0 比 | 1.0以下 | 1.2以上 |
| C26:0 濃度 | 1.0 μmol/L 以下 | 2.0 μmol/L 以上 |
| C26:0/C22:0 比 | 0.02以下 | 0.05以上 |
これらの基準値を超えた場合、確定診断のために遺伝子検査が推奨されます。
血液検査の流れ
血液検査によるVLCFAの測定は、以下の流れで行われます。
- 採血:少量の血液を採取
- ガスクロマトグラフィー・質量分析(GC-MS)または液体クロマトグラフィー(LC-MS)でVLCFAを分析
- 結果の解析:基準値と比較して異常の有無を判断
VLCFA測定と新生児スクリーニング
近年、新生児スクリーニングとしてVLCFA測定が導入されつつあり、生後早期の診断が可能になっています。
これにより、ALDの無症状の段階で治療介入ができるため、病気の進行を抑える可能性が高まっています。
血液検査でALDを早期発見する重要性
ALDは、早期診断によって進行を遅らせる治療が可能になります。
VLCFA測定は、簡便かつ精度の高い検査であり、特にALDの家族歴がある場合は、定期的に測定することが推奨されます。
もしALDが疑われる場合は、専門医の診察を受け、早期に血液検査を実施することが重要です。
MRI検査での脳白質の評価
磁気共鳴画像(MRI)検査は、副腎白質ジストロフィー(ALD)の診断および進行評価において最も重要な画像検査の一つです。
ALDの特徴である脳白質の異常を詳細に観察できるため、早期診断や治療方針の決定に不可欠です。
ALDの患者では、MRI検査で白質病変(脱髄)が確認されることが多く、特に小児大脳型ALDでは顕著な異常が認められます。
ALDにおける脳白質の異常とは?
ALDの病態は、ミエリン(神経を保護する白質)の破壊により、神経伝達が障害されることが特徴です。
MRI検査では、この白質の脱髄を視覚的に評価できます。
- 後頭葉・側頭葉の白質病変(小児大脳型ALDに多い)
- 脳梁の萎縮(神経機能の低下を示唆)
- 進行すると前頭葉にも拡大し、重度の神経症状を引き起こす
MRI検査による脳白質評価の目的
MRI検査は、以下の目的で実施されます。
- ALDの早期診断:白質病変の有無を確認
- 病状の進行評価:脱髄の進行度を経時的に追跡
- 治療効果のモニタリング:造血幹細胞移植後の経過観察
ALDのMRI画像の特徴
ALDのMRI画像では、以下のような特徴的な所見が見られます。
- T2強調画像:白質病変が高信号(白く映る)
- FLAIR画像:後頭葉や側頭葉の病変がより明瞭に観察される
- 造影剤(ガドリニウム)使用時:病変部の炎症を評価可能
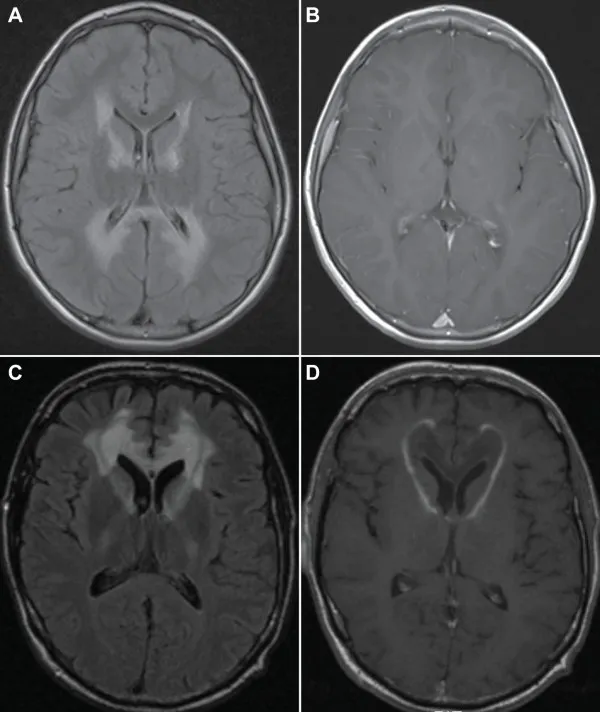

この領域は脳性ALDの約80%の症例で最初に侵される。
ガドリニウムを投与するとT1画像で縁が強調される(B)。
約20%の症例では脳性ALDの初期病変部位が前頭部白質であり、別の脳性ALD患者のFLAIR像(C)に示すように、
T1強調像(D)でガドリニウム投与後に顕著な縁の増強がみられる。
出典:ResearchGate
LoesスコアによるMRI評価
Loesスコアは、MRI画像を用いてALDの病変の広がりを数値化する指標です。
スコアが高いほど病気の進行度が大きく、予後の予測に役立ちます。
| Loesスコア | 病変の広がり | 予後 |
|---|---|---|
| 0~3 | 軽度 | 進行の可能性は低い |
| 4~9 | 中等度 | 神経症状が出現し始める |
| 10以上 | 重度 | 急速に進行し、重篤な障害を引き起こす |
MRI検査の実施タイミング
ALDの診断・経過観察のために、以下のタイミングでMRI検査が推奨されます。
- ALDの疑いがある場合(症状が現れたとき)
- 家族歴がある無症状の患者(定期スクリーニング)
- 造血幹細胞移植後の経過観察
MRI検査によるALDの早期発見の重要性
ALDは症状が出る前に白質病変が進行するため、MRI検査による早期発見が非常に重要です。
特に、家族歴がある場合は、定期的なMRI検査を受けることで、病気の進行を防ぐための適切な治療が可能になります。
遺伝子検査の重要性
副腎白質ジストロフィー(ALD)は、X染色体にあるABCD1遺伝子の変異によって発症する遺伝性疾患です。
遺伝子検査を行うことで、ALDの確定診断を行い、適切な治療や家族のリスク管理に役立てることができます。
遺伝子検査の目的
ALDの診断において、遺伝子検査は以下の目的で実施されます。
- 確定診断:血液検査やMRI検査でALDが疑われた場合、ABCD1遺伝子の変異を特定し、診断を確定する。
- 無症状キャリアの特定:ALDの家族歴がある女性のキャリア検査として利用。
- 発症リスクの評価:家族内での発症リスクを明確にし、早期治療の可能性を探る。
- 新生児スクリーニング:生後早期に診断し、症状が現れる前に適切な医療介入を行う。
遺伝子検査の方法
遺伝子検査は、血液または口腔粘膜のサンプルを採取し、ABCD1遺伝子の配列解析を行います。
現在、以下の方法が一般的に用いられています。
- サンガーシークエンス法:特定の遺伝子領域を詳細に解析。
- 次世代シークエンス(NGS):複数の遺伝子を一度に解析し、迅速な診断が可能。
- MLPA法:大きな遺伝子欠失や重複を検出。
遺伝子検査の結果とその意味
遺伝子検査の結果により、以下の3つのケースに分類されます。
| 検査結果 | 意味 |
|---|---|
| 陽性(変異あり) | ALDの発症リスクが高く、定期的な経過観察が必要。 |
| 陰性(変異なし) | ALDの可能性は低いが、家族内の発症リスクを考慮する必要がある。 |
| 不明(VUS: 意味不明変異) | 現時点では病的かどうか判断できず、追加の検査が必要。 |
遺伝子検査と遺伝カウンセリング
遺伝子検査の結果を正しく理解し、適切な対応を取るためには、遺伝カウンセリングが重要です。
特にALDは家族性の病気であるため、検査結果に基づき以下の点を検討することが推奨されます。
- キャリア女性のフォローアップ:発症リスクの評価と経過観察のスケジュール作成。
- 出生前診断や着床前診断の選択肢:家族計画における遺伝子検査の活用。
- 他の家族への検査の推奨:未診断の家族がいる場合、発症リスクを把握する。
遺伝子検査の早期実施の重要性
ALDは早期診断と治療介入が重要な疾患です。
遺伝子検査によってリスクを把握し、適切なフォローアップを行うことで、病気の進行を遅らせる可能性があります。
特に家族歴がある場合は、早めに専門医の診察を受け、遺伝子検査を検討することが推奨されます。
副腎白質ジストロフィーの治療法
造血幹細胞移植の適応と効果
造血幹細胞移植(Hematopoietic Stem Cell Transplantation, HSCT) は、副腎白質ジストロフィー(ALD)の進行を抑える唯一の根治的治療法として知られています。
特に小児大脳型ALDの患者に対しては、早期の適応判断が重要です。
造血幹細胞移植の適応基準
造血幹細胞移植は、以下の基準を満たす患者に適応されます。
- 小児大脳型ALDの早期段階(軽度の神経症状があるが、認知機能の大幅な低下がない)
- MRI検査でLoesスコアが10以下(白質病変の進行が限局的であること)
- 病気の進行スピードが早いと予測される場合
- 適合するドナーが見つかっている場合(同胞や骨髄バンクなど)
造血幹細胞移植は進行したALD患者には適応されないことが多いため、早期発見と移植のタイミングが極めて重要です。
造血幹細胞移植の方法
造血幹細胞移植には、以下の種類があります。
- 同種移植(Allogeneic HSCT):適合するドナーから造血幹細胞を提供
- 臍帯血移植:新生児の臍帯血に含まれる造血幹細胞を使用
- 自己移植(Autologous HSCT):遺伝子治療の一環として、患者自身の造血幹細胞を遺伝子改変後に移植
造血幹細胞移植の効果
造血幹細胞移植の最大の利点は、ALDの進行を抑制できることです。
特に小児大脳型ALDでは、移植によって脳の脱髄の進行を止めることが可能とされています。
- 神経症状の進行を抑制(発話・運動機能の維持)
- MRI検査で白質病変の進行停止が確認される
- 長期的な生存率の向上(適切な時期に移植を行えば、生存率が高まる)
ただし、すでに重度の神経症状がある場合は、移植の効果が限定的となるため、早期診断と適切なタイミングでの移植が鍵となります。
造血幹細胞移植のリスクと副作用
造血幹細胞移植には、以下のリスクが伴います。
- 移植片対宿主病(GVHD):ドナー細胞が患者の体を攻撃する自己免疫反応
- 感染症のリスク:免疫抑制状態になるため、細菌・ウイルス感染のリスクが高まる
- 拒絶反応:ドナー細胞が生着しないケースがある
- 化学療法による副作用:移植前の前処置として実施する化学療法の影響
造血幹細胞移植の成功率と予後
造血幹細胞移植の成功率は、移植のタイミングや患者の状態によって異なります。
| 移植のタイミング | 成功率 | 予後 |
|---|---|---|
| Loesスコア3未満 | 85%以上 | ほぼ正常な神経機能を維持 |
| Loesスコア4〜9 | 60〜75% | ある程度の神経症状が残る可能性 |
| Loesスコア10以上 | 30%以下 | 進行が抑制されにくい |
早期の移植ほど効果が高く、重度の神経症状が出る前に移植することが推奨されています。
造血幹細胞移植の適応を判断するための検査
造血幹細胞移植を行う前には、以下の検査が実施されます。
- MRI検査:白質病変の広がり(Loesスコア)を評価
- 血液検査:造血幹細胞の適合性を確認
- 遺伝子検査:ABCD1遺伝子変異を特定
- HLA適合性検査:ドナーとの適合性を評価
造血幹細胞移植の早期実施の重要性
造血幹細胞移植は、ALDの進行を抑える唯一の根本的な治療法ですが、適切な時期に行うことが成功の鍵となります。
ALDの家族歴がある場合や、MRI検査で白質病変が確認された場合は、早急に専門医と相談し、造血幹細胞移植の適応を検討することが重要です。
副腎皮質ホルモン補充療法の役割
副腎皮質ホルモン補充療法は、副腎白質ジストロフィー(ALD)に伴う副腎機能不全(アジソン病)の管理に不可欠な治療法です。
ALD患者の約90%が副腎皮質の機能低下を経験し、適切なホルモン補充を行わないと、倦怠感、低血圧、体重減少、色素沈着などの症状が悪化し、最終的には副腎クリーゼ(急性副腎不全)を引き起こす可能性があります。
副腎皮質ホルモンとは?
副腎皮質ホルモンには、以下の2種類があり、それぞれが重要な役割を果たします。
- コルチゾール(糖質コルチコイド):ストレス反応の調節、血糖値の維持、免疫機能の制御
- アルドステロン(鉱質コルチコイド):ナトリウムとカリウムのバランスを調整し、血圧を維持
ALD患者では、副腎機能が低下することでこれらのホルモンが不足し、全身の代謝や水分バランスが崩れるため、ホルモン補充が不可欠となります。
副腎皮質ホルモン補充療法の適応
以下のような症状が見られる場合、副腎皮質ホルモン補充療法の適応となります。
- 慢性的な疲労感や倦怠感
- 低血圧(起立性低血圧を含む)
- 体重減少や食欲不振
- 皮膚の色素沈着(特に関節部や口腔内)
- 低血糖発作
- 塩分の欲求増加
これらの症状が進行すると、副腎クリーゼを引き起こすリスクが高まり、緊急治療が必要になります。
副腎皮質ホルモン補充療法の治療薬
副腎皮質ホルモン補充療法では、以下の薬剤が使用されます。
| 薬剤名 | 作用 | 使用方法 |
|---|---|---|
| ヒドロコルチゾン(コートリル) | コルチゾールを補充し、ストレス反応を調整 | 通常1日2〜3回経口投与、ストレス時は増量 |
| フルドロコルチゾン(フロリネフ) | アルドステロンを補充し、血圧と電解質バランスを維持 | 1日1回経口投与 |
| デキサメタゾン | コルチゾール代替薬として使用されることがある | 長時間作用型で1日1回投与 |
副腎クリーゼを防ぐための管理
副腎機能不全があるALD患者では、ストレス時(感染症・手術・外傷など)に追加のホルモン補充が必要です。
副腎クリーゼを防ぐために、以下の点に注意する必要があります。
- 発熱や感染時にはヒドロコルチゾンの投与量を増やす
- 吐き気や嘔吐で薬を服用できない場合は、緊急時の注射薬を準備
- 低血圧や意識障害が現れた場合は、すぐに医療機関を受診
副腎皮質ホルモン補充療法の長期管理
副腎皮質ホルモン補充療法は生涯にわたる治療が必要ですが、適切な管理を行うことで、ALD患者の生活の質(QOL)を維持できます。
治療を継続する上で、以下の点が重要です。
- 定期的な血液検査(コルチゾールや電解質のモニタリング)
- ストレス時のホルモン補充計画を医師と相談
- 副作用(骨粗鬆症や高血圧)の管理
適切なホルモン補充と医療管理により、副腎機能不全の影響を最小限に抑え、健康的な生活を維持することが可能です。
副腎皮質ホルモン補充療法の早期開始の重要性
副腎機能不全は、早期に診断し、適切なホルモン補充を行うことで、重篤な合併症を防ぐことができます。
特にALDの家族歴がある場合は、定期的なホルモン検査を行い、発症前から予防的に治療を開始することが推奨されます。
早期診断と適切な治療を受けることで、副腎機能不全の症状をコントロールし、副腎クリーゼのリスクを最小限に抑えることが可能です。
ロレンツォオイル療法の可能性
ロレンツォオイル療法は、副腎白質ジストロフィー(ALD)の進行を抑えるために開発された食事療法の一つです。
ALDの発症に関与する極長鎖脂肪酸(VLCFA)の蓄積を低減することを目的としており、特に無症状または早期段階の患者に対する有効性が期待されています。
ロレンツォオイルとは?
ロレンツォオイル(Lorenzo’s Oil)は、オレイン酸(C18:1)とエルカ酸(C22:1)を主成分とする特殊なオイルで、VLCFAの合成を抑制する効果があります。
このオイルは、1980年代にALD患者であるロレンツォ・オドーネの両親によって開発され、現在でもALDの補助的治療として使用されています。
ロレンツォオイル療法のメカニズム
ALDの患者では、ペルオキシソームの機能異常により、極長鎖脂肪酸(VLCFA)が正常に分解されずに蓄積し、神経細胞や副腎にダメージを与えます。
- ロレンツォオイルに含まれるオレイン酸とエルカ酸が、VLCFAの合成を抑制
- 血液中のVLCFA濃度を低下させ、神経細胞への蓄積を減少
- 特に無症状のALD患者において、病気の進行を遅らせる可能性
ロレンツォオイル療法の適応
ロレンツォオイル療法は、以下の条件を満たす患者に適応されることが多いです。
- ALDの確定診断を受けたが、まだ症状がない無症候性患者
- 血液検査でVLCFA濃度が上昇している患者
- 造血幹細胞移植の適応外の患者
ただし、進行したALD患者に対してロレンツォオイル単独での治療効果は限定的であると考えられています。
ロレンツォオイル療法の臨床試験と効果
これまでに実施された研究では、ロレンツォオイルが無症状のALD患者におけるVLCFAの低下に寄与することが確認されています。
- 無症状の男児において、症状の発現を遅らせる可能性
- 血中VLCFA濃度を有意に低下
- ただし、すでに白質病変が進行している患者への効果は限定的
一部の研究では、ロレンツォオイルを使用しても病気の進行を完全に防ぐことはできないと報告されていますが、特定の患者群では一定の予防効果が期待されると考えられています。
ロレンツォオイル療法の副作用と注意点
ロレンツォオイル療法には、以下の副作用や注意点があります。
- 胃腸症状(下痢・腹痛)が発生することがある
- 血小板減少のリスクがあるため、定期的な血液検査が必要
- 長期間の使用による神経障害のリスクが指摘されている
そのため、ロレンツォオイル療法を実施する場合は、医師の指導のもとで慎重に管理することが重要です。
ロレンツォオイル療法の今後の可能性
現在、ロレンツォオイル療法の効果をさらに明確にするために、新たな臨床試験が進められています。
また、ロレンツォオイルと造血幹細胞移植や遺伝子治療との併用による効果の研究も進行中です。
今後の研究によって、ロレンツォオイルがALDの標準治療として確立される可能性があり、早期介入の選択肢の一つとして注目されています。
ロレンツォオイル療法を検討する際のポイント
ロレンツォオイル療法を検討する際には、以下の点を考慮することが重要です。
- 治療の対象となる患者群(無症候性ALD患者)を適切に選定
- VLCFAの低下効果を定期的に血液検査でモニタリング
- 単独治療ではなく、他の治療法と組み合わせる可能性を検討
ロレンツォオイル療法の早期導入の重要性
ロレンツォオイルは、無症状のALD患者に対する早期治療の選択肢として期待されています。
特に家族歴がある場合は、定期的なスクリーニングと早期の治療介入が重要です。
ただし、ロレンツォオイルのみでALDの進行を完全に防ぐことはできないため、他の治療法(造血幹細胞移植・遺伝子治療)との併用も検討する必要があります。
副腎白質ジストロフィーの予後と経過
病型別の進行パターン
副腎白質ジストロフィー(ALD)は、発症年齢や症状の進行度によって複数の病型に分類されます。
各病型には特徴的な進行パターンがあり、早期診断と適切な治療が患者の予後を大きく左右します。
小児大脳型ALDの進行パターン
小児大脳型ALD(Childhood Cerebral ALD, cALD)は、3~10歳の男児に発症し、急速に進行する神経症状を特徴とします。
- 初期症状として学習障害、注意力低下、行動変化が現れる
- 進行すると視力や聴力の低下、運動障害が発生
- 発症後2~5年以内に重度の神経障害や寝たきり状態に進行
- 適切な治療が行われない場合、数年以内に生命を脅かす状態に
MRI検査では、初期段階から後頭葉や側頭葉の白質病変が確認され、Loesスコアを用いた進行評価が重要となります。
副腎脊髄ニューロパチー(AMN)の進行パターン
副腎脊髄ニューロパチー(Adrenomyeloneuropathy, AMN)は、成人男性に発症し、ゆっくりと進行する神経症状が特徴です。
- 30~40代で歩行障害やバランス障害が出現
- 徐々に筋力低下、感覚異常、膀胱直腸障害が進行
- 長期間にわたり慢性的に進行し、最終的には車椅子が必要になるケースも
- 約30%の患者で小児大脳型ALDと類似した脳白質病変(炎症性ALD)が発生
副腎機能不全が進行すると副腎クリーゼのリスクが高まり、ホルモン補充療法が必要となります。
副腎不全型ALDの進行パターン
副腎不全型ALDは、神経症状を伴わずに副腎機能不全のみが発症するタイプです。
- 小児期に倦怠感、低血圧、色素沈着が出現
- 進行すると体重減少、食欲不振、脱水症状が顕著に
- ホルモン補充を行わない場合、副腎クリーゼを引き起こし命に関わる
早期診断によりヒドロコルチゾンやフルドロコルチゾンによるホルモン補充療法を開始することが重要です。
無症候性ALDの進行パターン
無症候性ALDは、ALDの遺伝子変異を持ちながらも、明確な症状がない状態を指します。
- 血液検査で極長鎖脂肪酸(VLCFA)の上昇が確認される
- 数年から数十年後に小児大脳型ALDやAMNに移行するリスクがある
- 定期的なMRI検査とVLCFAモニタリングが必要
無症候性の段階でロレンツォオイル療法や早期治療の検討を行うことが、進行を抑制する可能性があります。
病型別の進行を予測するための検査
ALDの進行を予測し、適切な治療を行うためには、以下の検査が重要です。
- 血液検査:VLCFAの測定
- MRI検査:白質病変の進行評価(Loesスコアの算出)
- 遺伝子検査:ABCD1遺伝子の変異解析
- 神経伝導検査:運動・感覚神経の機能評価
病型ごとの早期診断と治療の重要性
ALDの病型ごとの進行パターンを理解し、早期診断と適切な治療を行うことが、患者の予後を大きく改善する鍵となります。
特に小児大脳型ALDでは早期の造血幹細胞移植が推奨され、副腎機能不全型ではホルモン補充療法が必須となります。
定期的なMRI検査や血液検査を行い、ALDの進行を早期に発見し、適切な治療を行うことが最も重要です。
早期発見と治療の重要性
副腎白質ジストロフィー(ALD)は、進行性の神経変性疾患であり、早期に発見し適切な治療を行うことで、重篤な神経症状の発症を防ぐ可能性があります。
特に小児大脳型ALDでは、治療のタイミングが生存率やQOL(生活の質)に大きく影響します。
ALDの早期発見が重要な理由
ALDは発症初期には自覚症状が乏しく、気づかれにくい疾患ですが、次の理由から早期発見が極めて重要です。
- 小児大脳型ALDは急速に進行し、未治療の場合は数年以内に重篤な障害を引き起こす
- 造血幹細胞移植の効果は早期段階で最も高いため、白質病変が広がる前に治療を開始する必要がある
- 副腎機能不全を早期に診断し、ホルモン補充療法を行うことで致命的な副腎クリーゼを予防できる
- 家族歴がある場合、無症候性ALDのスクリーニングが可能であり、早期介入が可能になる
ALDの早期発見に有効な検査
ALDを早期に診断するためには、以下の検査が重要です。
- 血液検査:極長鎖脂肪酸(VLCFA)の測定によるスクリーニング
- MRI検査:脳白質病変の有無を確認(Loesスコアで進行度評価)
- 遺伝子検査:ABCD1遺伝子の変異を特定し、ALDの確定診断を行う
- 副腎機能検査:コルチゾールやACTHの測定により副腎機能不全を診断
ALDの早期治療の選択肢
早期に診断されたALD患者には、以下の治療法が適用されることがあります。
- 造血幹細胞移植(HSCT):小児大脳型ALDの進行を抑制する唯一の根治療法
- 副腎皮質ホルモン補充療法:副腎機能不全の管理に不可欠
- ロレンツォオイル療法:無症候性ALD患者におけるVLCFAの低減を目的とした食事療法
治療の効果は診断のタイミングに大きく依存するため、早期に適切な治療を開始することが不可欠です。
新生児スクリーニングによる早期発見の可能性
近年、多くの国で新生児スクリーニングにALDが追加されつつあり、無症状の段階で診断が可能になっています。
- 新生児期にVLCFA検査を行い、リスクのある児を特定
- 無症候性ALDが発見された場合、定期的なMRI検査と血液検査で経過観察
- 病変が進行する前に造血幹細胞移植などの適切な治療を実施
新生児スクリーニングの導入により、ALDの診断が症状が出る前に可能となり、患者の予後改善が期待されています。
早期発見・治療による予後の改善
早期発見と適切な治療により、ALD患者の予後は大きく改善されます。
| 診断・治療のタイミング | 予後 |
|---|---|
| 無症候性の段階で診断・造血幹細胞移植 | 90%以上が神経症状の進行を抑制できる |
| 小児大脳型ALDで早期治療開始(Loesスコア3未満) | 70~85%の患者で良好な予後 |
| 病気が進行した後に診断(Loesスコア10以上) | 進行を止めることが難しく、重篤な神経障害を伴う |
診断と治療のタイミングが予後を左右するため、早期のスクリーニングと継続的なモニタリングが重要です。
ALDの家族歴がある場合の対策
ALDはX連鎖性遺伝疾患であり、家族内で発症リスクが高い場合があります。
そのため、家族歴がある場合には、以下の対策を講じることが推奨されます。
- リスクのある家族に遺伝子検査を実施し、早期診断を確立
- 無症候性キャリアの特定と、定期的な検査による経過観察
- 発症リスクが高い場合は、出生前診断や着床前診断の選択肢を検討
早期発見と治療の重要性まとめ
副腎白質ジストロフィー(ALD)は進行性の神経疾患であり、早期に診断し、適切な治療を行うことで重篤な合併症を予防することが可能です。
- 新生児スクリーニングや遺伝子検査により、無症状の段階で診断が可能
- 造血幹細胞移植は、病気が進行する前に実施することで最大の効果を発揮
- 副腎機能不全の早期診断とホルモン補充療法が、致命的な副腎クリーゼを防ぐ
早期診断と適切な医療介入により、ALD患者の生活の質(QOL)を向上させ、病気の進行を抑えることが可能です。
副腎白質ジストロフィーの遺伝と家族計画
X連鎖性遺伝のメカニズム
X連鎖性遺伝は、X染色体上にある遺伝子の変異によって受け継がれる遺伝形式です。
副腎白質ジストロフィー(ALD)はX連鎖性劣性遺伝疾患に分類され、特に男性に多く発症します。
X連鎖性遺伝疾患では、男性と女性で遺伝の影響が異なるため、正しいメカニズムを理解することが重要です。
X連鎖性遺伝とは?
人間の性染色体は女性(XX)と男性(XY)で異なります。
- 女性(XX):X染色体を2本持つため、片方に異常があってももう片方のX染色体が補うことができる
- 男性(XY):X染色体を1本しか持たないため、異常があると発症しやすい
このため、X連鎖性遺伝疾患では男性の発症率が高く、女性はキャリア(保因者)として軽度の症状を示すことが多いです。
ALDにおけるX連鎖性遺伝の特徴
副腎白質ジストロフィー(ALD)はX染色体上のABCD1遺伝子の変異によって引き起こされます。
- 男性:変異を持つとほぼ100%発症
- 女性:通常は無症状または軽症だが、加齢に伴い神経症状が現れる場合がある
これは、女性が2本のX染色体を持っているため、もう片方の正常なX染色体が部分的に機能を補うことができるためです。
X連鎖性遺伝の遺伝パターン
ALDの遺伝は以下のように受け継がれます。
| 親の遺伝子型 | 子どもへの遺伝の可能性 |
|---|---|
| キャリアの母(X変異X) + 健康な父(XY) | 50%の確率で息子がALDを発症、50%の確率で娘がキャリア |
| ALDの父(X変異Y) + 健康な母(XX) | 100%の確率で娘がキャリア、息子には遺伝しない |
ALDの父親から息子には遺伝せず、母親がキャリアである場合に1/2の確率で息子が発症します。
X連鎖性遺伝疾患の診断と対策
X連鎖性遺伝疾患であるALDの発症リスクを評価するために、以下の検査が推奨されます。
- 遺伝子検査:ABCD1遺伝子の変異を特定
- 家族歴の確認:ALDの既往がある家族がいるかを調査
- 出生前診断:胎児の遺伝子検査を実施し、発症リスクを評価
- 新生児スクリーニング:生後すぐにALDの有無を診断
X連鎖性遺伝の理解と遺伝カウンセリング
ALDのようなX連鎖性遺伝疾患を持つ家族は、遺伝カウンセリングを受けることで、発症リスクの把握や将来の家族計画を立てることができます。
- キャリア女性の検査:発症リスクを評価し、定期的な経過観察を推奨
- 発症リスクのある家族への情報提供
- 適切な診断と早期治療の選択肢を検討
まとめ:X連鎖性遺伝のメカニズムを理解し、早期対策を
X連鎖性遺伝では、男性は発症しやすく、女性はキャリアとなる可能性が高いという特徴があります。
- ALDはX染色体のABCD1遺伝子の変異が原因
- 母親がキャリアの場合、息子の50%がALDを発症、娘の50%がキャリアとなる
- 遺伝子検査や新生児スクリーニングにより、早期診断が可能
家族歴がある場合は、遺伝カウンセリングを受け、適切な検査を実施することで、早期診断・早期治療が可能になります。
キャリア女性のリスクと対策
副腎白質ジストロフィー(ALD)はX連鎖性遺伝疾患であり、女性はキャリア(保因者)として遺伝子変異を持つことがあります。
一般的にキャリア女性は発症しないと考えられてきましたが、約30~50%の女性キャリアに神経症状が現れることが近年の研究で明らかになっています。
キャリア女性の発症リスク
病原性ABCD1バリアントを持つ女性は、成人期に症状を発症することが多いとされています。発症した場合、一般的に歩行障害や便失禁などの末梢神経障害が見られます。
症状の発生頻度は年齢とともに増加し、40歳未満の女性では20%未満であるのに対し、60歳を超えると90%近くに達することが報告されています。
一方で、副腎不全や脳への影響は女性では非常にまれです。最も精密な分析によると、線維芽細胞におけるX染色体不活性化と臨床症状のリスクとの間に明確な関連性は認められていません。しかし、過去の研究では異なる結論が示されていた例もあります。
女性におけるALDの特徴
- 30代以降に症状が現れることが多い
- 副腎機能不全はまれだが、神経症状が進行することがある
- 加齢とともに運動機能の低下が進行する可能性がある
キャリア女性に見られる主な症状
キャリア女性では、以下のような症状が見られることがあります。
- 歩行障害:軽度の痙性歩行、足のもつれ
- 感覚異常:下肢のしびれ、痛み
- 膀胱直腸障害:尿失禁、便秘
- 慢性的な疲労:倦怠感や筋力低下
- 軽度の認知機能障害:集中力や記憶力の低下
症状の進行は個人差が大きく、一部のキャリア女性では日常生活に支障をきたすケースもあるため、早期診断と定期的なフォローアップが重要です。
キャリア女性の診断方法
キャリア女性のALDリスクを特定するために、以下の検査が推奨されます。
- 遺伝子検査:ABCD1遺伝子変異の有無を確認
- 血液検査:極長鎖脂肪酸(VLCFA)の測定
- MRI検査:白質病変の有無を確認
- 神経伝導検査:下肢の神経障害を評価
キャリア女性のリスク管理と対策
キャリア女性がALDの影響を最小限に抑えるためには、以下の対策が有効です。
- 定期的な健康診断:血液検査やMRIを用いた経過観察
- 理学療法・運動療法:筋力低下を防ぎ、歩行障害を軽減
- 膀胱直腸障害の管理:専門医の診察を受け、適切な治療を実施
- ストレス管理:ストレスが症状を悪化させる可能性があるため、適切な休養をとる
家族計画と遺伝カウンセリング
キャリア女性が子どもを持つ場合、遺伝カウンセリングを受けることが推奨されます。
ALDはX連鎖性遺伝疾患のため、母親がキャリアの場合、50%の確率で息子が発症する可能性があります。
| 親の遺伝子型 | 子どもへの遺伝の可能性 |
|---|---|
| キャリア女性(X変異X) + 健康な男性(XY) | 50%の確率で息子がALDを発症、50%の確率で娘がキャリア |
| ALDの男性(X変異Y) + 健康な女性(XX) | 100%の確率で娘がキャリア、息子には遺伝しない |
キャリア女性が家族計画を考える際には、以下の選択肢を検討できます。
- 出生前診断:胎児の遺伝子検査を行い、発症リスクを評価
- 着床前診断(PGT):体外受精時に遺伝子検査を行い、ALDを持たない胚を選択
キャリア女性の早期対応の重要性
キャリア女性のALDリスクを正しく理解し、早期診断と適切な管理を行うことが重要です。
- 30歳以降に神経症状が現れる可能性があるため、定期的な検査を実施
- 歩行障害や感覚異常がある場合、早めに専門医の診察を受ける
- 家族計画を考える際は、遺伝カウンセリングを活用
キャリア女性は無症状のまま過ごすことが多いですが、神経症状が進行するリスクを軽減するためにも、早期からの対策が重要です。
遺伝カウンセリングの必要性
遺伝カウンセリングは、遺伝性疾患のリスクを持つ家族や個人に対して、発症リスクの評価、検査の選択肢、治療や予防策について専門的なアドバイスを提供する重要なプロセスです。
副腎白質ジストロフィー(ALD)はX連鎖性遺伝疾患であるため、家族内での遺伝の影響を正しく理解し、適切な対応をとることが不可欠です。
遺伝カウンセリングが必要な理由
遺伝カウンセリングは、以下のような理由から重要とされています。
- ALDの遺伝形式を理解し、家族内のリスクを明確にする
- 発症リスクのある家族に適切な検査を勧める
- 遺伝子検査の結果を正しく解釈し、今後の選択肢を考える
- 家族計画を立てる際のサポート(出生前診断や着床前診断の検討)
- 心理的なサポート(遺伝性疾患の診断に伴う精神的負担の軽減)
遺伝カウンセリングを受けることで、適切な情報を得て、最良の決定を行うことが可能になります。
遺伝カウンセリングを受けるべき対象者
以下のような状況に該当する方は、遺伝カウンセリングを受けることが推奨されます。
- ALDの家族歴がある(発症者またはキャリアの親族がいる)
- 自身または子どもがALDと診断された
- 妊娠を考えており、遺伝性疾患のリスクを評価したい
- 既に子どもを持つが、次の妊娠に向けてリスクを知りたい
遺伝カウンセリングの流れ
遺伝カウンセリングでは、以下のステップで情報提供と意思決定のサポートが行われます。
- 家族歴の確認:遺伝性疾患のリスクを特定
- 遺伝子検査の説明:必要な検査の種類や結果の意味を解説
- 検査結果の報告:ALDのリスク評価と今後の対応
- 家族計画や治療の選択肢の検討
- 心理的サポート:遺伝情報に対する不安の軽減
遺伝子検査と遺伝カウンセリングの関係
遺伝カウンセリングでは、遺伝子検査を受けるべきかどうかを判断し、検査結果に基づいた適切な対応を検討します。
ALDの診断においては、以下の検査が活用されます。
- ABCD1遺伝子検査:ALDの原因遺伝子の変異を特定
- 血液検査(VLCFA測定):極長鎖脂肪酸の異常蓄積を確認
- MRI検査:脳白質病変の進行を評価
家族計画と遺伝カウンセリング
遺伝カウンセリングは、家族計画を考える上でも重要な役割を果たします。
ALDの発症リスクがある場合、以下のような選択肢を検討することが可能です。
- 出生前診断:胎児の遺伝子検査を行い、発症リスクを評価
- 着床前診断(PGT):体外受精時に遺伝子検査を実施し、ALDを持たない胚を選択
これらの選択肢を適切に判断するためにも、専門家のアドバイスを受けることが重要です。
遺伝カウンセリングを受けるメリット
遺伝カウンセリングを受けることで、以下のようなメリットがあります。
- ALDの遺伝的リスクを正しく理解できる
- 発症リスクに応じた適切な検査を受けることができる
- 家族の健康管理や治療の選択肢について情報を得られる
- 将来の家族計画について安心して決断できる
- 心理的なサポートを受けることで不安を軽減できる
遺伝カウンセリングの早期受診の重要性
遺伝カウンセリングは、できるだけ早期に受けることが推奨されます。
早期に遺伝情報を把握することで、治療の選択肢を増やし、最適な対応を取ることが可能になります。
- 家族歴がある場合、早めに遺伝子検査を検討
- 妊娠を希望する際には、事前にリスク評価を行う
- 早期に診断を受けることで、治療の選択肢を確保
ALDのリスクを持つ家族は、専門医のサポートを受けながら、最適な選択をすることが重要です。

この記事の筆者:仲田洋美(医師)






