疾患に関係する遺伝子/染色体領域
疾患概要
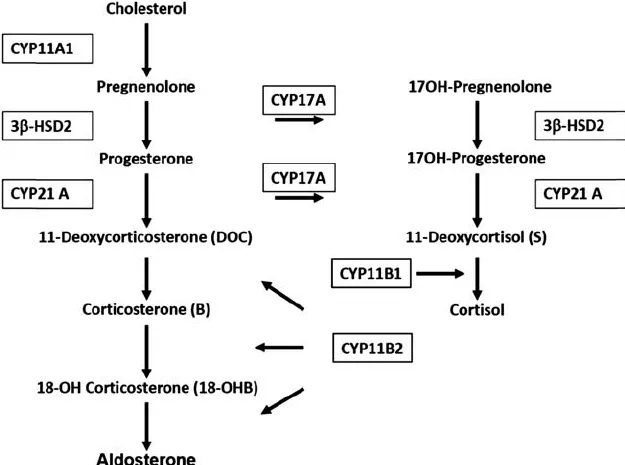
ステロイド17-モノオキシゲナーゼ、別名CYP17A1(遺伝子番号609300)は、17-α-ヒドロキシラーゼ活性と17,20-リアーゼ活性の両方を持つ酵素です。この酵素はステロイド合成経路における重要なステップを触媒し、特に性ホルモンと副腎ステロイドの生合成に関与しています。17-α-ヒドロキシラーゼ活性によって、プレグネノロンとプロゲステロンがそれぞれ17-α-ヒドロキシプレグネノロンと17-α-ヒドロキシプロゲステロンに変換され、これらはさらに性ホルモンへと変換される前駆体となります。17,20-リアーゼ活性は、これらの17-α-ヒドロキシ化されたステロイドをさらに分解し、アンドロゲンとなるデヒドロエピアンドロステロン(DHEA)やアンドロステンジオンへと変換します。
CYP17A1遺伝子の変異は、17-α-ヒドロキシラーゼ/17,20-リアーゼ欠損症という疾患を引き起こすことがあります。この疾患は、性ホルモンの合成障害により、性的発達の異常、生殖器の未発達、副腎機能不全などの多岐にわたる症状を示します。例えば、アンドロゲンの不足は男性の患者において女性化乳房や外性器の発達不全を引き起こし、女性の患者では二次性徴の欠如や無月経を引き起こすことがあります。また、この酵素の欠損は、副腎ステロイドの生合成にも影響を及ぼし、コルチゾールの不足やアルドステロンの過剰など、副腎皮質ホルモンの不均衡による症状を引き起こす可能性があります。
このようにCYP17A1遺伝子の変異によって引き起こされる表現型は、ステロイドホルモンの生合成経路におけるこの酵素の重要な役割を反映しています。そのため、CYP17A1遺伝子の機能不全に関連する疾患の診断と治療には、遺伝的検査やホルモン補充療法などが重要になります。
17α-ヒドロキシラーゼ/17,20-リアーゼ欠損症は、生殖腺(女性の場合は卵巣、男性の場合は精巣)と副腎の機能に影響を与える疾患です。これらの腺はホルモンの産生を司り、性的発達や生殖に重要な役割を果たします。この疾患は先天性副腎過形成症群に含まれ、ホルモンのバランスを崩し、性的発達や成熟を阻害します。
患者に見られるホルモンの不均衡は、高血圧、低カリウム血症、性的発達の異常などの特徴的な徴候や症状を引き起こします。症状の重症度は個人によって異なりますが、完全欠損症の方が重篤です。
女性患者では、外性器は正常ですが、子宮や卵巣などの内性器が未発達の状態です。完全欠損症の場合、二次性徴の発達が見られず、月経がありません(無月経)。部分欠損症の場合でも、二次性徴の発達は限定的で、月経は不規則または存在しないことが一般的です。どちらのタイプでも不妊となります。
男性患者では、性的発達に問題があり、外性器に異常が生じることがあります。最も重度の状態では、外性器が女性的で、通常は女性として育てられますが、内性器が発達していないため無月経となり、第二次性徴も現れません。精巣は存在しますが、正常な位置にないことがあります。完全欠損症の男性では、外性器が男女いずれにも該当しない外見を持つことがあり、部分欠損症の場合、陰茎が小さい、尿道口の位置異常、二分陰嚢などの特徴が見られることがあります。男性患者も不妊です。
この疾患の診断と管理は、性的発達の遅れ、不妊、および高血圧などの合併症に対処するために重要です。遺伝的検査を含む適切な医学的評価が必要であり、ホルモン補充療法などの治療が行われることがあります。
臨床的特徴
副腎17-ヒドロキシル化活性欠損症
副腎17-ヒドロキシル化活性の欠損症は、Biglieriら(1966年)によって初めて証明されました。この状態は、コルチコステロンとデオキシコルチコステロンの過剰産生により高血圧と低カリウム性アルカローシスを引き起こし、アルドステロン合成がほとんど認められない特徴があります。この症例の患者は正常な身長を持ち、無月経であった。保因者の血縁関係は認められなかったが、劣性遺伝の可能性が指摘されています。
Goldsmithら(1967年)は、17-α-水酸化の欠損が完全ではない可能性がある2人目の症例を報告しました。この女性患者は高血圧、原発性無月経、二次性徴の欠如を示しました。Mallin(1969年)は、2人の姉妹で見られる先天性副腎過形成を報告し、New(1970年)は男性患者で見られる偽両性具有を含む顕著な臨床特徴を報告しました。重度の高血圧や低カリウム血症は男性患者には見られませんでした。
Yazakiら(1982年)は、低カリウム血症性ミオパチー、原発性無月経、思春期発育の欠如を示す日本人女性(表現型)の症例を報告しました。腹腔内に精巣があり、核型は46,XYでした。グルココルチコイドの補充により、一部のホルモンレベルが正常化しました。
Scaroniら(1991年)は、性発達の欠如と低カリウム性高血圧を伴う3人の患者の家族を報告しました。デキサメタゾン治療によりカリウムが正常化しました。
Martinら(2003年)は、17α-ヒドロキシラーゼ/17,20-リアーゼ複合欠損症を有する11人の患者を報告し、基礎血清プロゲステロン測定が有用なマーカーであると結論づけました。
これらの報告は、17-ヒドロキシラーゼ/17,20-リアーゼ欠損症が原発性無月経、高血圧、低カリウム血症、性発達の異常など多様な臨床的特徴を持つことを示しています。治療にはグルココルチコイドの補充が有効であり、患者の症状の改善に寄与します。
17α-ヒドロキシラーゼ/17,20-リアーゼ欠損症
Oshiroら(1995)の研究では、32歳の日本人女性が複合型部分的17α-ヒドロキシラーゼ/17,20-リアーゼ欠損症の例として報告されました。この女性は、高血圧と無月経の症状で医療機関を訪れました。彼女は14歳で初潮を迎えましたが、20歳まで月経不順が続き、その後は無月経になりました。高血圧は20歳頃から知られていました。身体検査では、胸部の発育が不十分で、性器が幼児期の状態にあり、陰毛や腋毛の発育がありませんでした。CTスキャンにより、腫瘍のない両側副腎の過形成、子宮の低形成、および卵巣の萎縮が確認されました。さらに、彼女は低カリウム血症、代謝性アルカローシス、Trousseau徴候の陽性が確認されました。
一方、Yanaseら(1989)は、別の日本人女性のケースを報告しました。この女性は、17α-ヒドロキシラーゼ活性が正常の37%以下、17,20-リアーゼ活性が8%以下に低下していることが機能発現試験で明らかになりました。彼女は20歳で後頭部の頭痛を訴えて病院を訪れ、その際に高血圧と低カリウム血症が発見されました。月経不順があり、身体所見では乳房の発育不全と陰毛・腋毛の欠如が確認されました。Miuraら(1996)による追跡調査では、彼女がグルココルチコイド反応性高アルドステロン症と診断され、デキサメタゾン治療により28年間にわたって正常な血圧とカリウムレベルを維持していたことが報告されました。42歳で持続的な膣出血のため子宮全摘出術を受け、卵巣検体からは卵胞や黄体が検出されませんでした。45歳時には性ステロイドの減少とゴナドトロピンの増加が観察されました。この疾患は、加齢に伴う性腺機能の早期低下をもたらすことが示唆されました。
これらの報告から、CYP17A1遺伝子の変異による17α-ヒドロキシラーゼ/17,20-リアーゼ欠損症は、性ホルモンの産生障害に加え、ミネラルコルチコイドの過剰産生による高血圧や低カリウム血症など、多岐にわたる臨床的特徴を示すことが理解されます。これらの症状は、患者の残存する酵素活性のレベルによって異なり、症状の管理と治療には個別のアプローチが必要です。
孤立性17,20-リアーゼ欠損症
Gellerらの研究では、46,XY核型を持つ2人の患者で孤立性17,20-リアーゼ欠損症が確認されました。この疾患はCYP17A1遺伝子の異なるホモ接合体変異によって引き起こされます。最初の患者は性別の不明瞭さを示し、出生時に女性とされましたが、会陰部低空羂索や二股の陰嚢など、男性的な性器の特徴も持っていました。血清中のコルチゾール、電解質、血圧は正常であり、ゴナドトロピンの値も思春期前の正常範囲内でした。しかし、ヒト絨毛性ゴナドトロピン(hCG)による精巣刺激試験では、テストステロンや他のステロイドホルモンの応答が正常よりも低く、17,20-リアーゼの活性が欠如していることが示唆されました。
2人目の患者も出生時に生殖器のあいまいさが観察され、後に女性化乳房が発達しました。この患者もまた、hCG刺激試験においてテストステロンの生成が不十分であることが示され、17,20-リアーゼ欠損症の診断が支持されました。
Biason-Lauberらによる別の報告では、イスラエルの新生児男性患者が孤立性17,20-リアーゼ欠損症と一致するホルモンパターンを示し、CYP17A1遺伝子に複合ヘテロ接合変異があることが確認されました。しかし、Guptaらによるその後の研究では、これらの変異が診断と一致する活性を示すことはありませんでした。
Hershkovitzらは、上記の患者と他の3人の罹患男性近親者を再調査し、チトクロームP450酸化還元酵素(POR)の欠損症であるPOR遺伝子の変異を発見しました。この欠損症は孤立性17,20-リアーゼ欠損症として臨床的に現れることがあると結論付けられました。これは、孤立性17,20-リアーゼ欠損症がCYP17A1遺伝子の変異だけでなく、POR遺伝子の変異によっても引き起こされ得ることを示しています。
これらの報告は、孤立性17,20-リアーゼ欠損症の診断と管理において、遺伝子レベルでの詳細な検査が重要であることを強調しています。この疾患は性分化異常を引き起こし、患者の身体的、心理的な健康に影響を与えるため、正確な診断と適切な医療管理が必要です。
マッピング
遺伝
疾患が発症するためには、両親から受け継いだ遺伝子の両方で変異が存在する必要があります。両親がそれぞれ変異した遺伝子の1つのコピーを持っている場合、彼らは疾患の症状を示さないかもしれませんが、子供にその遺伝子の変異したコピーを2つ(ホモ接合体)伝えるリスクがあります。この場合、子供は疾患を発症する可能性があります。
常染色体劣性疾患の例としては、システィック・フィブローシス、鎌状赤血球症、タイ・サックス病などがあります。これらの疾患の場合、保因者の両親は症状を示さないかもしれませんが、両親から変異した遺伝子を受け継いだ子供は疾患の影響を受けることになります。常染色体劣性遺伝症の場合、遺伝子カウンセリングは、家族が遺伝的リスクを理解し、将来の子供に疾患が伝わる可能性について情報を得るために役立つことがあります。
頻度
17α-水酸化酵素/17,20-リアーゼ欠損症の発生頻度が世界中で約100万人に1人とされています。この疾患は、通常、常染色体劣性遺伝の形式を取ります。この情報を元に保因者の頻度を計算するためには、ハーディ・ワインバーグの平衡法則を使用します。この法則によると、疾患の頻度(q^2)がわかれば、保因者の頻度(2pq)を計算することができます。
疾患の発生頻度(q^2)は1/1,000,000 = 0.000001です。ここから、q(変異対立遺伝子の頻度)は√0.000001 = 0.001です。
ハーディ・ワインバーグの平衡では、p + q = 1ですから、p(正常対立遺伝子の頻度)は1 – q = 1 – 0.001 = 0.999です。
保因者の頻度(2pq)は次のように計算できます。
2pq = 2 * 0.999 * 0.001 = 0.001998、または約0.002です。
したがって、17α-水酸化酵素/17,20-リアーゼ欠損症の保因者の頻度は約0.2%、または約500人に1人と計算できます。これは理論的な計算であり、実際の頻度は地域や集団によって異なる可能性があることに注意してください。
原因
CYP17A1酵素は、ステロイドホルモン合成の過程で2つの重要な反応、すなわち17α-ヒドロキシラーゼ活性と17,20-リアーゼ活性を担っています。17α-ヒドロキシラーゼ活性はグルココルチコイドと性ホルモンの産生に関与し、17,20-リアーゼ活性は性ホルモンの産生に不可欠です。
この遺伝性疾患は、これらの酵素活性の欠如によって生じます。疾患は完全型と部分型に分けられ、完全型ではCYP17A1遺伝子の変異により酵素がほぼまたは完全に活性を失っています。一方、部分型では酵素活性は低下していますが、ある程度の活性が残っています。
17α-ヒドロキシラーゼ活性の欠如は、グルココルチコイドの産生に障害をもたらし、代わりにミネラルコルチコイドが過剰に産生されます。これが高血圧と低カリウム血症の原因となります。また、17,20-リアーゼ活性の欠如は性ホルモンの産生に影響を与え、性的発達や思春期の開始に障害をもたらします。
この疾患の診断は、臨床症状とホルモンレベルの検査、遺伝子検査により行われます。治療は、ホルモン補充療法を含め、患者の症状や欠損している酵素活性の種類に応じて行われます。ホルモン補充療法により、欠けているホルモンを補い、症状の改善や正常な発達を目指します。
分子遺伝学
Kagimotoらによる1988年の研究では、患者のCYP17A1遺伝子に4塩基の重複が同定され、これが両方の酵素活性の喪失につながることが発見されました。この変異は、17-α-ヒドロキシラーゼと17,20-リアーゼの両方の機能を失わせることにより、性ホルモン合成経路に影響を与えます。
一方、Gellerらによる1997年の研究では、ブラジルの小さな村に住む17,20-リアーゼ欠損症の患者2人からCYP17A1遺伝子に2種類のホモ接合体変異が同定されました。これらの変異体は、17-α-ヒドロキシラーゼ活性を保持しながら、17,20-リアーゼ活性を大幅に減少させました。これは、特定の変異が酵素の特定の機能に選択的に影響を及ぼすことができることを示しています。
Yanaseらによる1991年の報告によれば、17-α-ヒドロキシラーゼ欠損症は122例以上、17,20-リアーゼ欠損症は14例が報告されており、いくつかの症例では遺伝子内病変が同定されました。これらのデータは、疾患の遺伝的背景と臨床的多様性を示しています。
Martinらによる2003年の研究では、ブラジル人6家系11例の患者からCYP17A1遺伝子の複数の変異が同定されました。これらの変異は、患者における病態生理の理解を深め、特定の変異が特定の臨床的表現型にどのように関連しているかを示唆しています。
これらの研究は、17-α-ヒドロキシラーゼ/17,20-リアーゼ複合欠損症に関連する遺伝的変異の多様性と、それによって引き起こされる酵素活性の喪失が性ホルモン合成経路にどのように影響するかの理解を深めるものです。この知識は、疾患の診断、治療、および管理に重要な情報を提供します。
疾患の別名
17-alpha-hydroxylase-deficient congenital adrenal hyperplasia
Adrenal hyperplasia V
Combined 17 alpha-hydroxylase/17,20-lyase deficiency
Congenital adrenal hyperplasia due to 17-alpha-hydroxylase deficiency
Congenital adrenal hyperplasia type 5
Deficiency of steroid 17-alpha-monooxygenase
17α水酸化酵素欠損症
17α-ヒドロキシラーゼ欠損症先天性副腎過形成症
副腎過形成V
17α-ヒドロキシラーゼ/17,20-リアーゼ複合欠損症
17α-ヒドロキシラーゼ欠損症による先天性副腎過形成症
先天性副腎過形成5型
ステロイド17α-モノオキシゲナーゼ欠損症







