目次

BCAHH症候群は、KMT2D遺伝子のエクソン38・39に集中する特定のミスセンス変異によって引き起こされる、100万人に1人未満という超希少な先天性多発奇形症候群です。後鼻孔閉鎖・無乳頭症・難聴という特徴的な組み合わせを持ちながら、知的障害を伴わないという独特な表現型プロファイルが、早期の正確な診断を左右する重要な手がかりとなっています。
Q. BCAHH症候群とはどのような疾患ですか?まず結論だけ知りたいです
A. KMT2D遺伝子の特定領域(エクソン38・39)に生じるミスセンス変異によって引き起こされる、極めて稀な先天性多発奇形症候群です。鰓弓異常・後鼻孔閉鎖・無乳頭症・難聴・甲状腺機能低下症を主な特徴とし、知的障害を伴わないことが他の類似疾患と区別する重要なポイントです。
- ➤疾患の定義 → OMIM 620186、Orphanet ORPHA:589856、有病率100万人に1人未満
- ➤分子メカニズム → 歌舞伎症候群(ハプロ不全)とは異なる優性阻害・機能獲得メカニズム
- ➤主な症状 → 後鼻孔閉鎖(約78%)・無乳頭症・難聴(約89%)・甲状腺機能低下症
- ➤鑑別診断 → 歌舞伎症候群・CHARGE症候群との違いを詳解
- ➤診断・管理 → トリオ全エクソーム解析と集学的チーム医療の実際
1. BCAHH症候群とは:疾患の定義と歴史的背景
BCAHH症候群(OMIM 620186)は、その名称が5つの主要な臨床的特徴の頭文字から構成されています。Brachial arch abnormalities(鰓弓異常)、Choanal atresia(後鼻孔閉鎖)、Athelia(無乳頭症)、Hearing loss(難聴)、Hypothyroidism(甲状腺機能低下症)——この5症状の頭文字を取ってBCAHHと命名された、常染色体顕性遺伝形式をとる超希少疾患です。
💡 用語解説:常染色体顕性遺伝(じょうせんしょくたいけんせいいでん)
「常染色体」とは、性染色体(X・Y)以外の染色体のこと。「顕性(優性)」とは、2本の染色体のうちどちらか1本に変異があるだけで症状が現れることを意味します。BCAHH症候群では、変異した遺伝子を1つ持つだけで発症します。親から子へ遺伝する確率は理論上50%です。ただし、BCAHH症候群の多くはde novo(新生)変異——つまり両親には変異がなく、子どもで初めて生じた変異——によって発症するため、実際には親子間での遺伝は少ないと考えられています。
国際的な希少疾患データベースであるOrphanetには「ORPHA:589856」として登録されており、推定有病率は100万人に1人未満とされています。疾患が独立した症候群として確立されたのは比較的最近のことで、2020年にCuvertinoらとBaldridgeらによって独立して発表された二つの画期的な研究が、その出発点となりました。
歴史的な文脈として重要なのは、BCAHH症候群の原因遺伝子であるKMT2Dが、それ以前から歌舞伎症候群1型(Kabuki Syndrome 1 / KS1)の主要原因遺伝子として認識されていた点です。KS1は特徴的な顔貌・軽度から中等度の知的障害・胎児性指先隆起などを特徴とする疾患で、KMT2Dに変異が見つかると自動的に「歌舞伎症候群」と診断されてしまうリスクがありました。2020年の研究によって、同じKMT2D遺伝子であっても変異の部位と種類によって全く異なる症候群が生じることが初めて明らかになり、臨床遺伝学において重大なパラダイムシフトをもたらしました。
2. 原因遺伝子KMT2Dと分子病態メカニズム
BCAHH症候群の病態を理解するうえで核心となるのが、KMT2D遺伝子の特異な変異パターンとそれが引き起こすタンパク質レベルの構造変化です。このメカニズムは歌舞伎症候群とは根本的に異なります。
💡 用語解説:KMT2D遺伝子とは
KMT2D(Lysine Methyltransferase 2D)は、第12番染色体長腕(12q13.12)に位置する遺伝子で、ヒストンH3の4番目のリジン(H3K4)をメチル化する酵素をコードしています。H3K4のメチル化はクロマチン(DNA+タンパク質の複合体)を緩めて転写が活発な状態(オープンクロマチン)をつくる重要なエピジェネティックマーカーであり、胚発生・細胞分化・器官形成において欠かせない役割を担います。
💡 用語解説:エピジェネティクスとは
DNA塩基配列そのものを変えずに、遺伝子の「読まれ方(発現)」を制御する仕組みの総称です。DNAのメチル化やヒストンの修飾などが代表例。KMT2Dが触媒するH3K4メチル化は、特定の遺伝子を「オン」にするエピジェネティックスイッチとして機能します。この制御が乱れると、胎児発生の特定のプロセスが異常をきたします。
変異の「極端な局在性」——わずか40〜54アミノ酸の領域
歌舞伎症候群を引き起こすKMT2D変異が遺伝子全長にわたって広く分布しているのに対し、BCAHH症候群の変異は驚くほど狭い領域に集中しています。これまでに報告されたすべての病原性バリアントは、エクソン38・39にコードされるわずか40〜54アミノ酸の領域(Val3527〜Lys3583)にクラスター化しており、コイルドコイルドメイン(アミノ酸残基3562〜3614)の直前に位置しています。
エクソン38: c.10582C>G p.(Leu3528Val)、c.10595T>C p.(Ile3532Thr)、c.10621G>C p.(Ala3541Pro)、c.10625A>G p.(Tyr3542Cys)、c.10657T>G p.(Tyr3553Asp)
エクソン39: c.10745G>A p.(Arg3582Gln)、c.10744C>T p.(Arg3582Trp)、c.10763A>G p.(His3588Arg)
💡 用語解説:ミスセンス変異とデノボ変異
ミスセンス変異とは、DNA塩基が1つ変化することでアミノ酸が別の種類に置き換わるタイプの変異です。タンパク質の形が変わり、機能に影響を与えます。
デノボ(de novo)変異とは、両親の生殖細胞(精子・卵子)または受精直後に新たに生じた変異で、両親には同じ変異が存在しません。BCAHH症候群の多くはこのデノボ変異によって発症します。
なぜ歌舞伎症候群とは全く異なる疾患になるのか:優性阻害と機能獲得
歌舞伎症候群では、変異によってKMT2Dタンパク質の「量が半分に減る」ハプロ不全が病態の主体です。しかしBCAHH症候群では、エクソン38・39の特定ミスセンス変異によってタンパク質の二次構造(α-ヘリックスへの異常な転移)が物理的に乱れます。その結果、変異タンパク質は単に機能を失うのではなく、野生型タンパク質の正常な機能を積極的に妨害する——これが「優性阻害(ドミナントネガティブ)効果」または「機能獲得(Gain-of-Function)」と呼ばれるメカニズムです。
💡 用語解説:優性阻害(ドミナントネガティブ)効果とは
変異によって生じた異常タンパク質が、正常タンパク質の働きを「邪魔する」現象です。たとえば、あるタンパク質が複数集まって機能する複合体を形成する場合、1つの異常タンパク質が混入するだけで複合体全体の機能を損なわせることがあります。歌舞伎症候群の「量が足りない」状態(ハプロ不全)とは全く異なる病態メカニズムです。このメカニズムの違いが、同じKMT2D遺伝子の変異でありながらBCAHH症候群と歌舞伎症候群の臨床像が大きく異なる根本的な理由です。
さらに、ゲノム全体の末梢血DNAメチル化パターンを解析すると、BCAHH症候群患者群(エクソン38/39ミスセンス変異コホート)は、歌舞伎症候群患者群とは統計的に有意に(p < 0.001)異なる独自のメチル化プロファイル(エピジェネティック・シグネチャー)を示すことが確認されています。この分子レベルの差異が、BCAHH症候群を独立した疾患として分類する強力な根拠となっています。
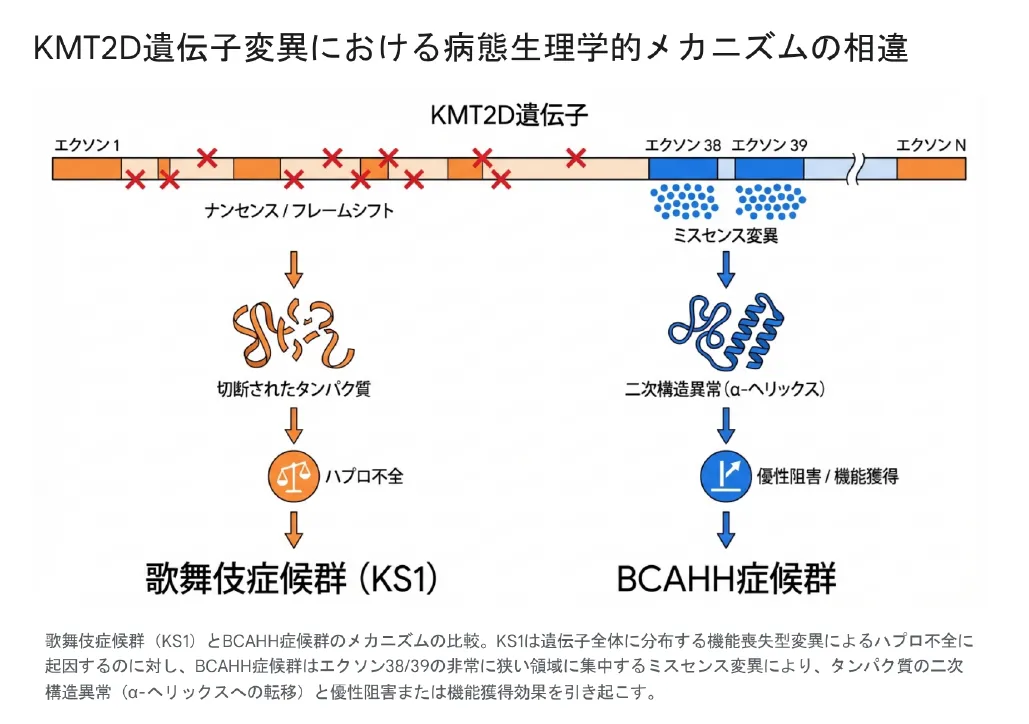
KMT2D遺伝子全体に分布するナンセンス・フレームシフト変異はハプロ不全を介して歌舞伎症候群を引き起こすのに対し、エクソン38/39の特定ミスセンス変異はタンパク質の二次構造を乱し、優性阻害または機能獲得効果によってBCAHH症候群を引き起こす。
3. 主な症状と表現型スペクトラム
BCAHH症候群の臨床像は、複数臓器にまたがる重篤かつ特徴的な先天奇形の集合体です。Cuvertinoら(7家系9名)とBaldridgeら(4名)のコホート研究によって、主要な症状とその出現頻度が明らかにされています。
🦻 耳鼻咽喉・聴覚
- 難聴(伝音性・感音性・混合性):89〜90%
- 鰓弓異常(鰓裂嚢胞・頸部瘻孔など):78%
- 外耳奇形(小耳症・外耳道閉鎖症など):43〜67%
👃 呼吸器・鼻腔
- 後鼻孔閉鎖(両側性または片側性):71〜78%
- 重症間質性肺疾患(ILD):一部重症例に発症
⚕️ 内分泌・成長
- 甲状腺機能異常:52〜67%
- 思春期遅発または欠如:高頻度
- 極端な低身長・成長障害:38%以上
- 副甲状腺機能低下症:約19%
👁️ 皮膚・眼・その他
- 無乳頭症または乳頭形成不全:43〜67%
- 涙管異常(形成不全・閉塞):57〜78%
- 先天性心疾患(ASDなど):一部
- 知的障害:0%(欠如)
💡 用語解説:後鼻孔閉鎖(こうびこうへいさ)
鼻腔の後方開口部(後鼻孔)が骨組織または膜組織によって閉鎖または狭窄した状態です。胎生期(通常35〜38日目)における口鼻膜の破裂不全が原因と考えられています。新生児は鼻呼吸に絶対的に依存しているため、両側性の場合は出生直後から致死的な呼吸窮迫とチアノーゼを引き起こし、緊急の気道確保と外科的介入が必要になります。CHARGE症候群でも高頻度に見られますが、BCAHH症候群との鑑別が重要です。
💡 用語解説:無乳頭症(アセリア / Athelia)
乳頭が先天的に欠損している、または極めて低形成な状態です。男女ともに認められます。歌舞伎症候群では女性患者の約30%で乳房の早期発育が見られるのとは対照的に、BCAHH症候群では無乳頭症が43〜67%に認められます。この「無乳頭症の存在」はBCAHH症候群を他の多くの先天性多発奇形症候群から鑑別する最大の臨床的手がかりの一つです。

知的障害が「ゼロ」という特筆すべき事実
BCAHH症候群を他の多発奇形症候群から際立てる最も重要な特徴は、知的障害の欠如(0%)です。歌舞伎症候群やCHARGE症候群では中等度から重度の知的障害がほぼ普遍的に見られるのとは全く対照的です。Cuvertinoらのコホート(9名)では知的障害を持つ患者は1名も存在しませんでした。ただし一部患者では言語表出の遅れや運動発達の遅れが報告されており、認知機能は保たれていても発達支援は重要です。
また、BCAHH症候群では歌舞伎症候群の鑑別上重要な陰性所見として、胎児性指先隆起(指先の膨らみ)・口蓋裂・構造的腎異常が認められないことも挙げられます。これらが「ない」という情報も診断の方向性を絞る重要な手がかりです。
4. 鑑別診断:歌舞伎症候群・CHARGE症候群との違い
BCAHH症候群の診断における最大の課題は、他の先天性多発奇形症候群との著しい表現型のオーバーラップです。特に新生児期には、複合症状から別の症候群と誤診されるリスクが高くなります。
歌舞伎症候群1型(KS1)との鑑別
BCAHHにあってKS1にない特徴:
後鼻孔閉鎖(KS1では稀)、無乳頭症(KS1では報告なし)
KS1にあってBCAHHにない特徴:
知的障害、胎児性指先隆起、口蓋裂、外側下眼瞼の外反と長い眼裂という特徴的顔貌
CHARGE症候群との鑑別
注意点:後鼻孔閉鎖・難聴・心疾患・成長障害などが重なり、CHARGE症候群の診断基準を満たしてしまうケースが多々あります。半規管の異常(CHARGEに特徴的)が見られた例もあります。
鑑別のポイント:無乳頭症の存在と知的障害の欠如、そしてCHD7遺伝子の陰性とKMT2Dエクソン38/39変異の同定によって明確に区別されます。
ヨハンソン・ブリザード症候群との鑑別
無乳頭症・鼻翼の低形成・難聴の組み合わせから鑑別診断に挙がります。
鑑別のポイント:原因遺伝子がUBR1であり、重篤な膵外分泌不全や頭皮欠損はBCAHHには認められません。遺伝子パネル検査によって除外可能です。
AIを用いた深層学習顔貌解析(Face2Geneなど)を用いた客観的評価でも、BCAHH患者の顔面ゲシュタルトは歌舞伎症候群の顔貌モデルから統計的に有意に区別されます(AUC = 0.918)。BCAHHにおける顔貌の特徴はより多様であり、顔面の非対称性・眼距開離・鼻翼の著しい低形成・テント状の薄い上唇・下垂した鼻柱などが報告されていますが、歌舞伎症候群の「決め手となる顔貌」は存在しません。
5. 診断基準と遺伝子検査の進め方
BCAHH症候群は2020年に独立した疾患として提唱されたばかりであり、世界的に統一された診断基準はまだ確立されていません。しかし最新の文献に基づき、以下の階層的な診断アプローチが推奨されています。
臨床的レッドフラッグ:これが揃ったらBCAHHを強く疑う
💡 BCAHH症候群を疑うべき主要所見の組み合わせ
- ➤後鼻孔閉鎖(Choanal atresia)
- ➤無乳頭症(Athelia)または著明な乳頭形成不全
- ➤難聴または外耳・鰓弓奇形
- ➤原因不明の低カルシウム血症(副甲状腺機能低下症)または甲状腺機能低下症
これらのレッドフラッグに加え、口蓋裂の欠如・胎児性指先隆起の欠如・知的障害の欠如・顔貌がKS1典型例と異なることが、診断の方向性をCHARGE症候群や歌舞伎症候群からBCAHHへと転換させる重要な手がかりとなります。
分子遺伝学的検査:トリオ全エクソームシーケンスが最強のツール
💡 用語解説:トリオ全エクソームシーケンス(Trio-WES)
WES(Whole Exome Sequencing)とは、遺伝子のタンパク質をコードする領域(エクソン)全体を網羅的に解析する次世代シーケンス手法です。「トリオ」とは患者本人だけでなく、両親も含めた3名で同時解析することを指します。デノボ変異(両親にはなく子どもに新しく生じた変異)を効率よく検出できるため、BCAHH症候群のように多くがデノボ変異で生じる疾患の診断に特に有効です。単一遺伝子検査や限られた遺伝子パネルでは見逃すリスクがあるため、トリオWESが推奨されます。
診断における重要な落とし穴は、バリアントの解釈にあります。KMT2Dに変異が見つかった場合、自動的に「歌舞伎症候群」と診断されてしまうリスクがあります。臨床遺伝専門医とバイオインフォマティクス担当者は、同定されたミスセンス変異がエクソン38または39の高度保存領域(アミノ酸3527〜3583付近)に該当するかどうかを厳密に評価しなければなりません。
この領域の変異(例:p.Ile3532Thrやp.Leu3528Valなど)は、従来「意義不明のバリアント(VUS)」と分類されていたとしても、ACMGの最新基準と報告論文と照合することで「病的(Pathogenic)」または「病的である可能性が高い(Likely Pathogenic)」に再分類できるケースが存在します。過去に歌舞伎症候群と診断されていた患者がBCAHH症候群と再診断される事例もあることに注意が必要です。
補助診断:DNAメチル化シグネチャー解析とAI顔貌解析
💡 用語解説:DNAメチル化シグネチャー解析(エピジェネティックプロファイリング)
末梢血DNAのメチル化パターン(どのゲノム部位がメチル化されているか)を網羅的に解析することで、疾患固有の「メチル化の指紋(シグネチャー)」を同定する手法です。EpiSignなどの解析ツールによって、BCAHH症候群に特有のメチル化プロファイルが確認されれば、VUSとして分類されていたKMT2Dエクソン38/39ミスセンス変異の病原性を生化学的に証明し、診断を確定的なものにすることができます。歌舞伎症候群との統計的に有意な分離(p<0.001)がすでに確認されており、次世代の診断補助ツールとして注目されています。
6. 治療と長期管理プロトコル
BCAHH症候群は多臓器にわたる障害を引き起こすため、小児科・耳鼻咽喉科・内分泌科・呼吸器科・循環器科・臨床遺伝科からなる集学的チーム医療体制の構築が不可欠です。
新生児期の気道確保:最優先の緊急対応
両側性の後鼻孔閉鎖は出生直後に致死的な呼吸不全を引き起こすため、直ちに経口エアウェイの挿入または気管内挿管により気道を確保し、速やかに耳鼻咽喉科医による経鼻的内視鏡下開通術(Endoscopic choanoplasty)を計画します。術後も肉芽形成による再狭窄のリスクがあるため、顔面の成長が終了するまでの長期フォローアップが強く推奨されます。また一部患者では重症間質性肺疾患(ILD)を発症するため、持続的な低酸素血症やばち指の出現に注意し、定期的な胸部高分解能CT(HRCT)評価が必要です。
内分泌管理:複数腺性の異常に対する包括的対応
甲状腺機能低下症
2021年発表のERN(欧州参照ネットワーク)コンセンサスガイドラインに準拠し、診断後速やかにレボチロキシン(Levothyroxine)補充療法を開始。TSH・遊離T4の頻回モニタリングと厳格な用量調整が神経発達保護に直結します。
副甲状腺機能低下症
新生児期の原因不明の重症低カルシウム血症に注意。ESE(欧州内分泌学会)ガイドラインに準じ、カルシウムと活性型ビタミンD製剤を補充。低カルシウム血症クリーゼやテタニーの予防と、過剰補充による腎石灰化の回避を同時に管理します。
低身長・思春期遅発
成長ホルモン分泌不全の有無を内分泌学的に評価。思春期の欠如・遅発に対しては骨年齢を定期評価し、適切な時期に性腺ステロイド製剤による思春期導入療法の必要性を検討します。心理社会的サポートも並行して重要です。
聴覚・眼科・その他の管理
難聴が約89%に見られるため、生後早期の聴性脳幹反応(ABR)・耳音響放射(OAE)検査が必須です。外耳道閉鎖や小耳症を伴うことが多く、従来の気導補聴器の装着が物理的に困難な場合が多いため、骨導補聴器(BAHA)の導入や人工内耳埋め込みのタイミングを専門医と協議します。涙管閉塞や形成不全による反復性涙嚢炎に対しては、保存的治療が無効な場合に涙管ブジーやシリコンチューブ留置術を検討します。心房中隔欠損症などの先天性心疾患が潜在することもあり、確定診断時に心エコー検査による評価を行います。
7. 遺伝カウンセリングの意義
BCAHH症候群の確定診断後、家族への丁寧な遺伝カウンセリングが必要です。遺伝カウンセリングで扱われる主な内容は以下の通りです。
- ➤遺伝形式と再発リスクの説明:多くのケースはデノボ変異であり、両親への遺伝は認められません。ただし常染色体顕性遺伝のため、患者本人が子どもを持つ場合の遺伝確率は理論上50%です。生殖細胞モザイクの可能性も除外できないため、次子の出生前診断についても検討が必要です。
- ➤予後情報の提供:知的障害を伴わないという事実は、患者の教育・社会参加・自立に向けた長期的な見通しを立てるうえで、家族にとって非常に重要な希望の根拠となります。
- ➤出生前診断の選択肢:次子を望む場合、絨毛検査・羊水検査による出生前遺伝子診断が選択肢として存在します。既知の変異が同定されている場合は確実な診断が可能です。
- ➤心理的サポートと情報収集の継続:疾患の希少性から国内外の患者レジストリや患者団体の情報が限られています。長期的な自然歴の蓄積に向けた医療機関との連携継続が重要です。
8. よくある誤解
誤解①「KMT2D変異=歌舞伎症候群」
KMT2Dに変異が見つかっても自動的に歌舞伎症候群とは言えません。エクソン38・39の特定ミスセンス変異はBCAHH症候群を引き起こします。変異の部位と種類の精密な解釈が必要です。
誤解②「知的障害がないから軽症」
知的障害がないことはBCAHH症候群の特徴であり「軽症」を意味しません。後鼻孔閉鎖・重症間質性肺疾患・低カルシウム血症クリーゼなど、生命に直接関わる重篤な合併症を有します。
誤解③「CHARGE症候群に似ているからCHARGEだ」
初期にCHARGE症候群の診断基準を満たしてしまうケースは実際に報告されています。最終的にはCHD7陰性・KMT2Dエクソン38/39変異の同定によって鑑別されます。トリオWESが重要です。
誤解④「親も同じ変異があるはず」
BCAHH症候群の多くはデノボ(新生)変異であり、両親には同じ変異が存在しないことがほとんどです。「両親が健康だから遺伝ではない」という誤解が診断を遅らせることがあります。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少疾患の診断・遺伝カウンセリングについて
BCAHH症候群をはじめとする希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] NCBI MedGen. Choanal atresia-athelia-hypothyroidism-delayed puberty-short stature syndrome (Concept Id: C4479027). [NCBI MedGen]
- [2] Orphanet. Choanal atresia-athelia-hypothyroidism-delayed puberty-short stature syndrome. ORPHA:589856. [Orphanet]
- [3] Cuvertino S, et al. A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet Med. 2020;22(5):867-877. [PMC7200597]
- [4] Baldridge D, et al. Phenotypic expansion of KMT2D-related disorder: Beyond Kabuki syndrome. Am J Med Genet A. 2020;182(6):1506-1512. [PMC7295006]
- [5] Kabuki and CHARGE syndromes: overlapping symptoms and diagnostic challenges. Einstein (São Paulo). 2025. [PMC11869784]
- [6] Bedwell JR, et al. Management of Choanal Atresia: National Recommendations with a Comprehensive Literature Review. Children (Basel). 2023;10(1):91. [MDPI]
- [7] Léger J, et al. Congenital Hypothyroidism: A 2020–2021 Consensus Guidelines Update. Thyroid. 2022;32(4):395-420. [PMC8001676]
- [8] OMIM #620186. BCAHH Syndrome. Johns Hopkins University. [OMIM]






