疾患に関係する遺伝子/染色体領域
疾患概要
オーメン症候群は、重症複合型免疫不全(SCID)を伴う常染色体劣性遺伝疾患です。この疾患は、紅皮症(皮膚の激しい発赤)、肝脾腫(肝臓や脾臓の肥大)、リンパ節症(リンパ節の肥大)、および脱毛症(髪の脱落)などの特徴を持ちます。B細胞はほとんど存在せず、T細胞数は通常から増加しており、T細胞は頻繁に活性化されていますが、T細胞受容体(TCR)のレパートリーは限られています。このため、感染に対する効果的な免疫応答ができません(Egeら、2005年)。
また、家族性組織球性網状血球症の別の形態は、10q22染色体上のパーフォリン-1遺伝子(PRF1)の突然変異によって引き起こされることが知られています。
オーメン症候群は、遺伝性の免疫不全症で、免疫システムが正常に機能せず、細菌やウイルス、真菌に対する防御力がほとんどない状態を引き起こします。この疾患は重度複合免疫不全症(SCID)の一種で、患者は繰り返し深刻な感染症にかかりやすく、生命を脅かすこともあります。オーメン症候群の乳児は、肺炎や慢性下痢にかかりやすく、これらの感染を引き起こす微生物は通常、健康な人には無害な日和見感染菌です。
この疾患に特徴的なのは、免疫不全に加え、免疫システムが自分の体の組織や臓器を攻撃する自己免疫反応が起こることです。その結果、皮膚の激しい発赤(紅皮症)、脱毛(脱毛症)、肝臓や脾臓の肥大(肝脾腫)が見られます。また、リンパ節や胸腺などのリンパ組織が異常に肥大します。
治療を受けない場合、オーメン症候群の子供は通常1~2歳までしか生存できませんが、免疫機能を回復させる治療、特に骨髄移植などが有効とされています。
遺伝的不均一性
臨床的特徴
Omenn症候群は、免疫不全を伴う重症複合免疫不全症(SCID)の一種であり、重篤な症状が特徴です。Omenn(1965年)は、アイルランド系アメリカ人の近親婚家族において、好酸球増加症を伴う網内系疾患を初めて報告しました。その後、Barthら(1972年)は、これを独立した疾患として確認しました。患者には、紅皮症(皮膚の発赤)、肝脾腫(肝臓や脾臓の肥大)、リンパ節腫大、脱毛症などが見られ、B細胞がほとんど存在せず、T細胞数は正常から増加していますが、T細胞受容体(TCR)のレパートリーが限定されているため、感染や自己免疫反応を引き起こしやすくなります。
Egeら(2005年)は、5か月の乳児がOmenn症候群の症状を示し、敗血症、発育不全、肝脾腫、紅皮症、脱毛症などが現れた症例を報告しました。患者の皮膚には魚鱗癬様の鱗屑と紅色丘疹が認められ、好酸球の増加とT細胞の異常増殖が観察されました。免疫抑制療法により症状が一時的に改善した後、HLA半合体型の母親からの幹細胞移植が成功し、免疫機能が正常化しました。この患者は、IgEレベルの上昇や好酸球増加を示す、T+、B-、NK+のSCID免疫表現型を持つ典型的なOmenn症候群の例でした。
この症候群は、T細胞の自己攻撃(自己免疫)が強く、皮膚、腸、肝臓、脾臓などに深刻な影響を与えます。また、Omenn症候群は移植片対宿主病(GVHD)に類似した病態を示すことがあり、T細胞の異常な増殖と反応が見られます。
生化学的特徴
マッピング
遺伝
この症状は、常染色体劣性遺伝パターンで遺伝します。つまり、各細胞の遺伝子の両コピーに変異があることを意味します。常染色体劣性遺伝の症状を持つ個人の両親は、それぞれ変異遺伝子のコピーを1つずつ保有していますが、通常はその症状の兆候や症状は示しません。
頻度
全体として、SCIDのさまざまな形態は、新生児75,000~100,000人に1人の割合で発症すると推定されています。オーメン症候群の正確な発症率は不明です。
原因
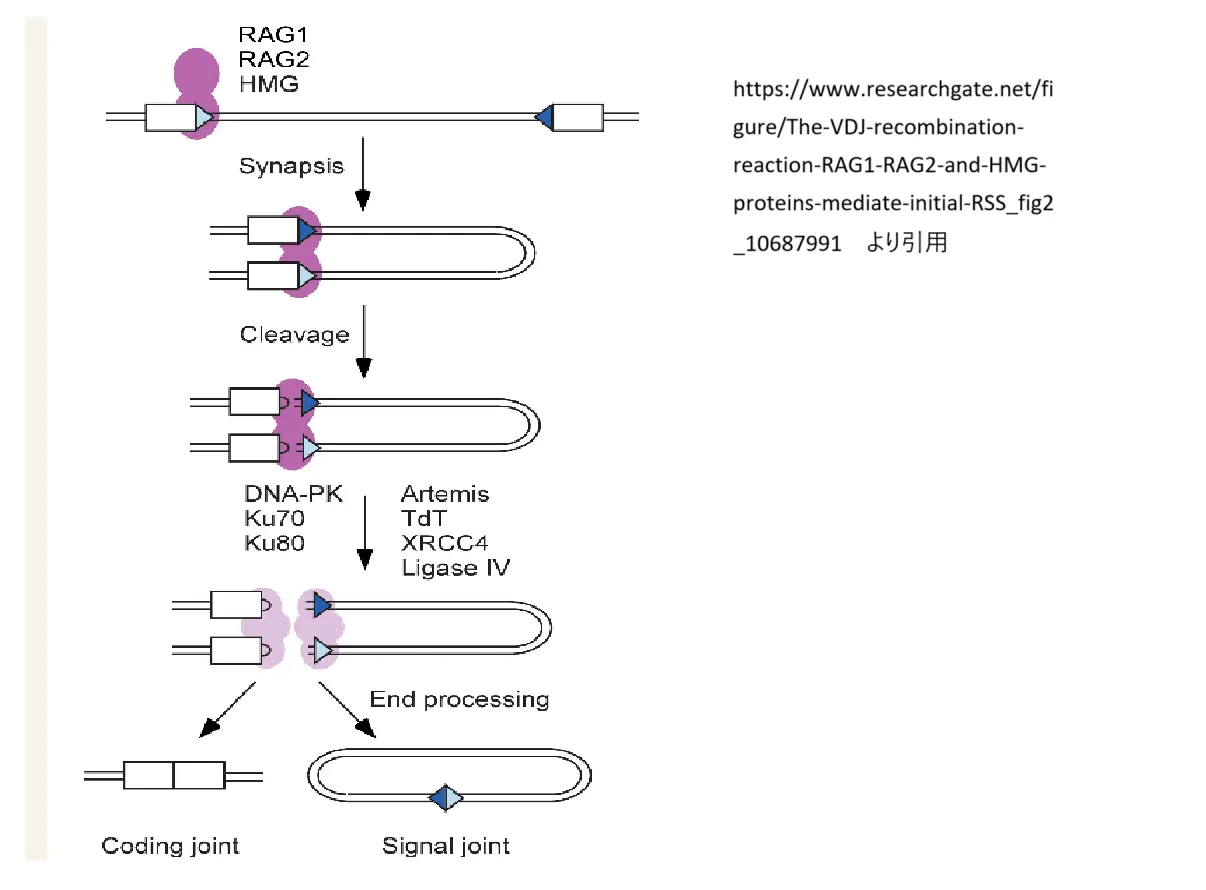
オーメン症候群は、主にRAG1およびRAG2遺伝子の突然変異が原因で発症します。これらの遺伝子は、免疫システムにおいて重要な役割を果たすB細胞とT細胞という2種類のリンパ球に関与しています。RAG1とRAG2のタンパク質は、B細胞とT細胞の表面にあるタンパク質の多様性を高め、これにより異物を認識しやすくします。この多様性がないと、細胞は外来の病原体を効果的に認識し、攻撃することができなくなります。
オーメン症候群を引き起こすRAG1およびRAG2の突然変異は、これらのタンパク質の機能を著しく低下させ、B細胞とT細胞の表面タンパク質の多様性を大きく制限します。その結果、これらのリンパ球が病原体を認識して対抗する能力が低下し、頻繁に感染症を引き起こします。特にT細胞は数が正常でも、機能が異常であり、自己免疫反応を引き起こして自分の組織を攻撃してしまうため、オーメン症候群では自己免疫疾患が併発します。
Egeら(2005年)は、オーメン症候群の症状を示した兄弟3人について報告しています。2人はすでにこの症候群により亡くなり、3人目の兄弟もオーメン症候群と診断されました。遺伝子解析により、この家族の3人目の兄弟はDCLRE1C/Artemis遺伝子における2つの異なる変異(605988.0012および605988.0013)を持つ複合ヘテロ接合型であることが確認されました。この遺伝子変異は、アルテミスタンパク質の機能に影響を与え、免疫システムの異常やオーメン症候群の発症につながると考えられています。
治療・臨床管理
Gomezら(1995年)は、1980年から1989年の間に治療したOmenn症候群の患者9名に対する同種骨髄移植(BMT)の結果を報告しました。彼らの研究では、移植前に非経口栄養と免疫抑制療法を実施することを条件として、HLA一致または半合致のBMTのいずれもがOmenn症候群を治癒する可能性があることが示されました。この結果により、骨髄移植がOmenn症候群の効果的な治療法であると結論づけられました。
病因
Cavadiniら(2005年)は、リアルタイムPCRおよび免疫組織化学法を用いて、Omenn症候群患者2名とT-, B-, NK+ SCID患者1名の胸腺におけるAIRE(自己免疫調節因子)の発現を調査しました。その結果、正常な対照群と比較して、これらの免疫不全患者ではAIRE mRNAおよびタンパク質の発現が大幅に減少していることが確認されました。また、AIREに関連する自己抗原であるインスリンやチトクロームP450 1A2、脂肪酸結合タンパクのmRNAは、免疫不全患者では検出されませんでした。
この研究の結果、Cavadiniらは、AIRE発現の欠損が重度の免疫不全症においてT細胞の異常な発達を引き起こす可能性があると結論付けました。Omenn症候群では、少数の残存するT細胞クローンが胸腺でのネガティブセレクション(自己反応性T細胞の排除)を回避し、末梢で異常に増殖することで、自己免疫反応を引き起こす可能性が示唆されました。
分子遺伝学
Villaら(1998年)は、Omenn症候群の患者において、RAG1(179615.0001-179615.0013)またはRAG2(179616.0003; 179616.0004)遺伝子にミスセンス変異があることを報告しました。これらの変異により、RAG1とRAG2タンパク質の活性が部分的に低下します。特に、RAG1のホメオドメイン内でアミノ酸置換が起こるとDNA結合活性が低下し、他の変異ではRAG1/RAG2相互作用の効率が低下しました。この発見は、Omenn症候群における免疫不全がV(D)J組み換えの効率低下に起因することを示しています。
さらに、Corneoら(2001年)は、T-、B-SCID患者においてRAG2遺伝子の2つの変異(179616.0002; 179616.0008)の複合ヘテロ接合を特定しました。この遺伝子型はOmenn症候群の同胞でも確認されました。また、無関係な2人のT-、B-SCID患者でも、RAG1遺伝子の変異(179615.0010; 179615.0015)が見つかり、これらの変異はOmenn症候群患者にも見られました。著者らは、Omenn症候群の発症には、これらの変異に加えてさらなる因子が関与していると結論づけました。
Egeら(2005年)は、Omenn症候群の症状で兄弟2人が死亡し、3人目の兄弟がDCLRE1C/Artemis遺伝子における複合ヘテロ接合変異(605988.0012; 605988.0013)を持っていた家族について報告しています。この変異もOmenn症候群に関連しています。
動物モデル
Khiongら(2007年)は、Rag1遺伝子に自然発生した突然変異を持つマウスを特徴づけ、これらのマウスがOmenn症候群のモデルとして適していると結論づけました。このモデルは、Omenn症候群の病態メカニズムを理解するための重要なツールとなりました。
また、Marrellaら(2007年)は、Omenn症候群患者に見られるRag2遺伝子のarg229-to-gln変異(R229Q; 179616.0002)を内在性Rag2に置換したノックインマウスモデルを作成しました。このマウスは、ヒトOmenn症候群に見られるほとんどの症状を再現しており、研究者たちは、このモデルがヒトの疾患理解や治療研究に役立つと結論づけました。
疾患の別名
Familial reticuloendotheliosis 家族性細網内皮症
Histiocytic medullary reticulosis 組織球性髄質細網症
Omenn’s syndrome オーメン症候群
参考文献

この記事の監修・執筆者:仲田 洋美
(臨床遺伝専門医/がん薬物療法専門医/総合内科専門医)
ミネルバクリニック院長。1995年に医師免許を取得後、
臨床遺伝学・内科学・腫瘍学を軸に診療を続けてきました。
のべ10万人以上のご家族の意思決定と向き合ってきた臨床遺伝専門医です。
出生前診断(NIPT・確定検査・遺伝カウンセリング)においては、
検査結果の数値そのものだけでなく、
「結果をどう受け止め、どう生きるか」までを医療の責任と捉え、
一貫した遺伝カウンセリングと医学的支援を行っています。
ハイティーンの時期にベルギーで過ごし、
日本人として異文化の中で生活した経験があります。
価値観や宗教観、医療への向き合い方が国や文化によって異なることを体感しました。
この経験は現在の診療においても、
「医学的に正しいこと」と「その人にとって受け止められること」の両立を考える姿勢の基盤となっています。
また、初めての妊娠・出産で一卵性双生児を妊娠し、
36週6日で一人を死産した経験があります。
その出来事は、妊娠・出産が女性の心身に与える影響の大きさ、
そして「トラウマ」となり得る体験の重みを深く考える契機となりました。
現在は、女性を妊娠・出産のトラウマから守る医療を使命の一つとし、
出生前診断や遺伝カウンセリングに取り組んでいます。
出生前診断は単なる検査ではなく、
家族の未来に関わる重要な意思決定です。
年齢や統計だけで判断するのではなく、
医学的根拠と心理的支援の両面から、
ご家族が後悔の少ない選択をできるよう伴走することを大切にしています。
日本人類遺伝学会認定 臨床遺伝専門医/日本内科学会認定 総合内科専門医/
日本臨床腫瘍学会認定 がん薬物療法専門医。
2025年には APAC地域における出生前検査分野のリーダーとして国際的評価を受け、
複数の海外メディア・専門誌で特集掲載されました。