目次

AHCY欠損症(S-アデノシルホモシステインヒドロラーゼ欠損症)は、世界でわずか15〜20例しか詳細な臨床経過が報告されていない、有病率100万人に1人未満の超希少な神経代謝疾患です。単一の酵素(SAHH)の欠損が全身のメチル化反応を根底から破綻させ、精神運動発達遅滞・重篤なミオパチー・肝障害・特異な血液凝固異常という三主徴をもたらします。診断の鍵となるのはメチオニン濃度ではなく、血漿中のSAMとSAHの定量測定です。
Q. AHCY欠損症とはどのような疾患ですか?まず結論だけ知りたいです
A. AHCY遺伝子の先天的変異によりS-アデノシルホモシステインヒドロラーゼ(SAHH)が欠損し、有害な代謝産物SAH(S-アデノシルホモシステイン)が全身に異常蓄積する常染色体劣性遺伝の超希少疾患です。SAHが体内40種以上のメチル基転移酵素を強力に阻害することで、精神運動発達遅滞・ミオパチー・肝障害・血液凝固異常という多臓器に及ぶ重篤な症状を引き起こします。
- ➤疾患の定義 → Orphanet ORPHA:88618、有病率100万人に1人未満、世界で15〜20例のみ
- ➤分子メカニズム → SAH蓄積(正常値の最大190倍)→ メチル基転移酵素阻害 → DNAエピジェネティック破綻
- ➤主な症状 → 精神運動発達遅滞・ミオパチー(筋緊張低下)・肝障害・血液凝固異常(特に第VII因子低下)
- ➤鑑別診断 → MAT I/III欠損症・ホモシスチン尿症・GNMT欠損症との違いを詳解
- ➤診断・治療 → メチオニン値は初期に正常のことも。SAM/SAH測定が鍵。肝移植が根治的選択肢として注目
1. AHCY欠損症とは:疾患の定義と疫学
AHCY欠損症(正式名:S-アデノシルホモシステインヒドロラーゼ欠損症を伴う高メチオニン血症)は、AHCY遺伝子にコードされる酵素「S-アデノシルホモシステインヒドロラーゼ(SAHH)」の先天的な活性低下または欠損によって引き起こされる、常染色体劣性遺伝の神経代謝疾患です。
💡 用語解説:常染色体劣性遺伝(じょうせんしょくたいれっせいいでん)
「常染色体」とは、性別を決める性染色体(X・Y)以外の46本のうち44本の染色体のこと。「劣性(潜性)」とは、両方の染色体に変異が揃ったときだけ発症することを意味します。1本だけ変異があっても通常は発症しませんが、変異を持つ親を「保因者(キャリア)」と呼びます。両親が2人ともキャリアの場合、妊娠ごとに25%の確率でお子さんが発症します。
国際希少疾患データベース「Orphanet」には ORPHA:88618 として登録されており、推定有病率は100万人に1人未満とされています。医学文献で詳細な臨床経過が報告されている症例は世界全体でわずか15〜20例程度に過ぎない、まさに「ウルトラオーファン疾患(超希少疾患)」です。
多くの症例は乳児期に発症し、①精神運動発達遅滞、②著明な筋緊張低下を伴うミオパチー、③特異な血液凝固異常を伴う肝機能障害という「三主徴」を呈します。ただし、その重症度には非常に広いスペクトラムがあります。最重症例では胎児水腫として胎児期から発症し乳児期早期に死亡するケースがある一方、成人期まで比較的軽微な症状で生存する例や、26歳になって初めて確定診断された例も報告されています。
2. 原因遺伝子AHCYと分子病態メカニズム
AHCY欠損症の病態を理解するには、体内のメチオニン代謝サイクルと、SAHHが果たす不可欠な役割を知ることが重要です。AHCY遺伝子の詳細についてはAHCYの遺伝子詳細ページもあわせてご覧ください(Aで始まる遺伝子一覧)。
SAHHとメチオニン代謝サイクルの核心
メチオニンは生体においてタンパク質合成の素材であるだけでなく、細胞のエピジェネティック制御の要となる必須アミノ酸です。体内ではメチオニンからSAM(S-アデノシルメチオニン)が合成され、DNAやヒストンなど40種以上の重要な分子にメチル基を付与します(メチル基転移反応)。このメチル基が転移された後に残る副産物がSAH(S-アデノシルホモシステイン)です。
💡 用語解説:SAM/SAH比(メチル化指数)
SAM(メチル基を供与する物質)とSAH(供与後に生じる副産物)の濃度比を「メチル化指数」と呼びます。この比が高いほど、細胞が活発にメチル化反応を行える状態であることを示します。AHCY欠損症ではSAHが最大190倍に蓄積し、SAMも最大50倍に増加することでこの比が著しく低下し、全身のメチル化機能が崩壊します。
この生化学プロセスにおいて、SAHを処理する唯一の酵素がSAHHです。SAHHは、SAHをアデノシン(プリン系ヌクレオシド)とホモシステインに分解します。この酵素が先天的に欠損すると、SAHが細胞内・血中に異常蓄積し始めます。
SAHは単なる廃棄物ではなく、体内のほぼすべてのメチル基転移酵素に対する強力な阻害剤として作用します。SAHが蓄積すると上流のSAMも代謝されずに溜まり、さらにその上流のメチオニンも蓄積するという悪循環が生じ、「高メチオニン血症」として現れます。
なお、SAHHの反応で生成されるアデノシンは、プリン系ヌクレオシドとして下図のようなイノシン酸(IMP)合成経路とも深くつながっています。SAHH欠損によるアデノシンの代謝異常は、核酸代謝全般にも影響を及ぼすことが知られています。
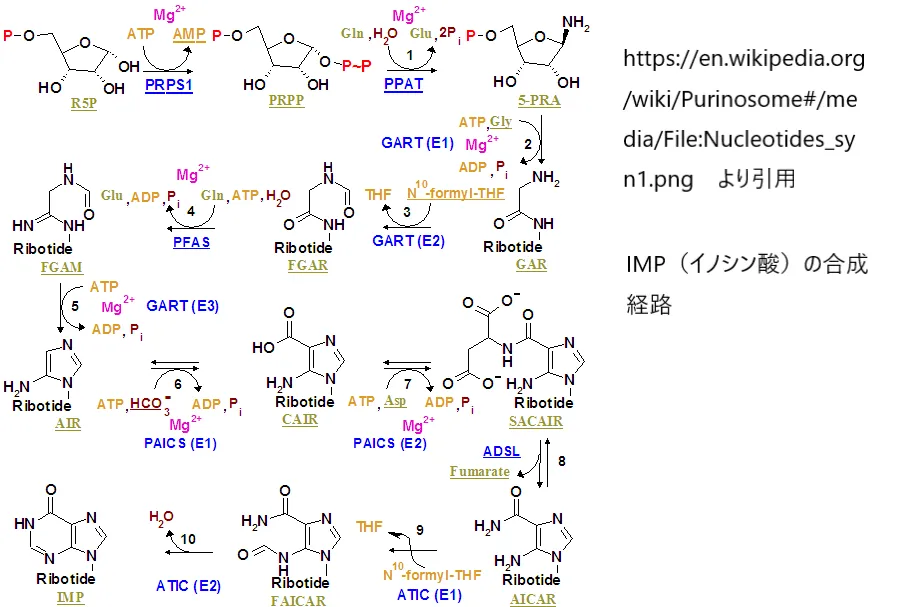
https://en.wikipedia.org/wiki/Purinosome#/media/File:Nucleotides_syn1.png
より引用
イノシン酸(IMP)を起点としてAMPが合成されるプリンヌクレオチド合成経路。SAHHが触媒するSAH→アデノシン+ホモシステインの反応はこのプリン代謝とも連絡しており、SAHH欠損はアデノシン代謝にも影響を与える。
SAH蓄積がもたらす二大カスケード障害
SAHによるメチル基転移酵素の広範な阻害は、特に以下の2つの重要な経路に致命的な打撃を与えます。
💪 クレアチン合成の停止
GAMT(グアニジノ酢酸-N-メチルトランスフェラーゼ)が阻害されることで、筋肉の主要エネルギー貯蔵物質であるクレアチンの合成が停止します。これが重篤なミオパチーと筋緊張低下の直接的な原因です。
🧠 髄鞘形成の障害
PEMT(ホスファチジルエタノールアミン-N-メチルトランスフェラーゼ)が阻害され、ホスファチジルコリン(細胞膜・ミエリン鞘の主成分)が著減します。これが神経髄鞘化遅延と白質萎縮の根本原因です。
💡 用語解説:エピジェネティクスとDNAメチル化
DNAの塩基配列そのものを変えずに、遺伝子の「読まれ方」を制御する仕組みを「エピジェネティクス」と呼びます。DNAのシトシン塩基にメチル基(CH₃)が付くと、その遺伝子は読まれにくく(発現が抑制)なります。AHCY欠損症では本来この「メチル化」が低下するはずですが、実際には代償的なフィードバック機構により逆に高メチル化が生じると考えられています。
SAHがDNAメチルトランスフェラーゼ(DNMT1)を阻害するなら、DNA「低メチル化」が起きると予想されます。しかし実際に患者の白血球DNAを解析すると、逆に広範なDNA高メチル化が観察されるという生物学的パラドックスが明らかになっています。この高メチル化の最も重大な臨床的帰結が、血液凝固第VII因子遺伝子(F7)のプロモーター領域の異常メチル化です。F7プロモーターにある転写因子の結合部位がメチル化によってブロックされることで、第VII因子の産生が根本から抑制され、患者特有の重篤な凝固障害が生じます。
AHCY遺伝子:染色体上の位置と報告されている主な変異
AHCY遺伝子は染色体20q11.22に位置し、生物進化において極めて高度に保存された遺伝子です。細菌から哺乳類まで構造と機能が維持されており、モデル生物での完全なノックアウトは胚発生の初期段階で致死的となります。したがって、ヒトで報告されているすべての症例は酵素活性を部分的に維持したミスセンス変異やナンセンス変異の組み合わせです。
🧬 主な病原性バリアントと臨床的特徴
- p.R49C:四量体(ホロ酵素)形成を著しく阻害→タンパク質の急速な分解・活性喪失
- p.D86G:酵素の立体構造を不安定化。p.R49Cとの複合ヘテロ接合では胎児水腫・乳児期早期死亡という最重症の転帰
- p.Y143C:重度のミオパチー・発達遅滞。肝臓でのSAHH活性が対照の約3%まで低下
- p.A89V:p.Y143Cとの複合ヘテロ接合で26歳まで生存した症例。中等度の知的障害(全検査IQ 64)
- p.G71S:生後8ヶ月まではメチオニン・ホモシステインが完全に正常だった特異な例
AHCY欠損症診断時のSAM・SAH上昇倍率(正常値=1倍)
SAM
S-アデノシルメチオニン
SAH
S-アデノシルホモシステイン
報告された患者群における初期評価時の正常値に対する上昇倍率(中央値)。初期乳児期においてもSAHの著明な蓄積が診断の強力な指標となる(Data sources: NCBI PMC)
3. 主な症状と多臓器への影響
AHCY欠損症の臨床的特徴は単一臓器にとどまらず、極めて多岐にわたります。その発症時期は、胎児期(胎児水腫・先天性脳構造異常)から成人期の遅発性肝不全まで、驚くほど広範なスペクトラムを示します。
🧠 中枢神経系
- 全体的な精神運動発達遅滞
- 脳の髄鞘化遅延(ミエリン鞘形成不全)
- 脳梁の低形成・小脳・橋の低形成
- 白質萎縮・脳室拡大・小頭症
- 内斜視・眼球運動障害
- 重度の知的障害・言語発達障害
💪 筋骨格系
- 著明な筋緊張低下(フロッピーインファント)
- 頭部コントロール不良・起立・歩行困難
- クレアチンキナーゼ(CK)の著増
- 筋ジストロフィー様の筋病理所見
- 進行性の呼吸筋障害リスク
🫀 肝臓・消化器
- ALT・ASTの持続的上昇(肝炎様所見)
- 超音波:肝実質のエコー輝度低下
- 肝細胞癌(HCC)発症リスクの上昇
- 成人期に肝不全として初発する例も
🩸 血液凝固異常
- プロトロンビン時間(PT)の著明な延長
- 第VII因子・第II因子・第IX因子・第X因子の著減
- アンチトロンビンIII・プロテインS・プラスミノーゲン低下
- 致死的な消化管・頭蓋内出血リスク
💡 用語解説:フロッピーインファント(筋緊張低下の乳児)
生まれた赤ちゃんの全身の筋肉がぐにゃぐにゃと軟らかく、抱き上げると手足がだらんとした状態を指します。英語で「フロッピー(ぐにゃぐにゃした)」と表現します。AHCY欠損症では筋肉内のクレアチン(エネルギー貯蔵物質)の著しい枯渇がこの状態を引き起こします。頭を自分で持ち上げることができない、うつ伏せで腕立て状態になれないなどの症状として現れます。
本疾患の臨床像で特に重要なのは、胎児期からの最重症例では胎児水腫として発症するケースがあることです。このような重篤な胎児期発症に関しては、非免疫性胎児水腫(NIHF)包括的NGSパネルや胎児水腫の検査との関連で検討されることがあります。
その他、顔貌異常・毛髪の質感変化・白内障・心筋症・感覚運動ニューロパチーなども報告されており、SAHHという単一酵素の欠損が、いかに広範な組織発生プログラムを狂わせるかを示しています。また少なくとも1例で肝細胞癌(HCC)が確定診断されており、長期的な発癌リスク管理も重要な課題です。

4. 鑑別診断:他の高メチオニン血症との違い
新生児スクリーニングで「高メチオニン血症」として検出された場合、複数の疾患を正確に鑑別することが治療方針の決定に不可欠です。AHCY欠損症と他の高メチオニン血症の鑑別には、メチオニン値だけでなく、SAM・SAH・総ホモシステイン(tHcy)の組み合わせパターンが決定的です。
MAT I/III欠損症(MAT1A遺伝子)との鑑別
生化学的相違:MAT I/III欠損症ではSAMが低下〜正常、SAHは正常。対してAHCY欠損症ではSAM・SAHともに著増。
臨床的相違:MAT I/III欠損症の多くは無症状の良性疾患。ミオパチー・凝固異常はない。
ホモシスチン尿症(CBS欠損症)との鑑別
生化学的相違:CBS欠損症ではtHcy(総ホモシステイン)が著増。AHCY欠損症ではtHcyは正常〜微増。
臨床的相違:CBS欠損症では水晶体脱臼・マルファン症候群様の骨格異常・血栓塞栓症が特徴的。ミオパチーはない。
GNMT欠損症(GNMT遺伝子)との鑑別
生化学的相違:GNMT欠損症ではSAMが上昇するがSAHは正常〜軽度上昇のみ。tHcyは正常。
臨床的相違:軽度の肝酵素上昇や肝腫大。重篤なミオパチーや凝固異常は認められない。
I型チロシン血症(FAH遺伝子)との鑑別
生化学的相違:チロシン著増・サクシニルアセトンの尿中排泄が特徴。高メチオニン血症は二次性(肝不全に伴う)。
臨床的相違:進行性の肝不全・腎尿細管機能障害・くる病。ニチシノン(NTBC)治療が有効。
5. 診断:バイオマーカーと遺伝子検査
AHCY欠損症の診断において最大の落とし穴は、メチオニン濃度への過信です。新生児スクリーニングではメチオニン値のみが測定されますが、報告された12例中6例で初期メチオニン値が正常でした。p.G71S変異を持つ患児では生後8ヶ月を超えるまでメチオニン・ホモシステインともに正常値を示し続けた例すら存在します。
一次バイオマーカー:SAMとSAHの定量評価
本疾患に対して最も信頼性の高い特異的バイオマーカーは、血漿中のSAH(S-アデノシルホモシステイン)とSAM(S-アデノシルメチオニン)の定量評価です。すべての報告症例で病初期から例外なく劇的な増加が確認されています。同時に総ホモシステイン(tHcy)が正常〜微増に留まることを確認することが、ホモシスチン尿症(tHcy著増)との決定的な鑑別点となります。
💡 用語解説:全エクソームシーケンス(WES)
WES(Whole Exome Sequencing / 全エクソームシーケンス)とは、遺伝子のタンパク質をコードする領域(エクソン)すべてを網羅的に解析する次世代シーケンス手法です。ヒトゲノム全体の約1.5%しか占めませんが、既知の病的変異の約85%がこの領域に集中しているため、希少疾患の原因探索に非常に強力です。AHCY欠損症の確定診断には、WESまたはAHCY遺伝子を含む代謝疾患パネルによる塩基配列解析が推奨されます。
確定診断のアルゴリズム
- Step 1:血漿アミノ酸分析(メチオニン)+血漿総ホモシステイン(tHcy)測定
- Step 2:臨床所見(ミオパチー・CK著増・肝障害・凝固異常)があればメチオニン正常でもAHCY欠損症を除外しない
- Step 3:血漿SAM・SAH定量評価→ SAH著増・SAM著増・tHcy正常〜微増の組み合わせで強く疑う
- Step 4:AHCY遺伝子の塩基配列解析(WESまたは代謝疾患パネル)で両アレルの病的バリアントを同定→確定診断
- Step 5(補助):SAHH酵素活性の直接アッセイ(肝臓・線維芽細胞・赤血球)
関連する遺伝子検査として、ミネルバクリニックではコバラミン・ホモシステイン・メチオニン遺伝子検査や総合代謝NGSパネル、また鑑別疾患のカバーにはメチルマロン酸尿症・ホモシスチン尿症NGSパネルを提供しています。
6. 治療・長期管理
現在のところ、AHCY欠損症のエピジェネティックな破綻を完全に修復するような確立された根治的治療は存在しません。治療の焦点は、代謝負荷を軽減し病態の進行を遅らせることに向けられています。
保存的治療:メチオニン制限食と補充療法の組み合わせ
🥗 メチオニン制限食
天然タンパク質を制限し、メチオニンフリーフォーミュラを補充。1日あたりメチオニン摂取量を15〜40mg/kgという狭い治療域に管理します。血漿メチオニン・SAM・SAHを大幅に低下させることが可能。
💊 クレアチン補充療法
300〜375mg/kg/日のクレアチン経口補充により、枯渇した筋肉のエネルギー代謝プールを直接補填。筋緊張低下の改善とミオパチーの進行抑制を目的とします。
🧪 ホスファチジルコリン補充
150〜200mg/kg/日の補充。細胞膜とミエリン鞘(髄鞘)の主要材料を提供し、中枢神経系の髄鞘形成をサポートします。
⚠️ SAMeは禁忌
MAT I/III欠損症では有効なSAMe(S-アデノシルメチオニン)補充は、AHCY欠損症では絶対に禁忌です。すでにSAMが病的に蓄積しているため、SAMeを摂取すると代謝異常がさらに悪化します。
保存的治療の限界
限界② CK(ミオパチー指標)・肝トランスアミナーゼの異常はほとんどの症例で改善しない
限界③ 白血球DNAの広範な高メチル化状態は、34ヶ月の食事療法後も正常化されなかった
限界④ 精神運動発達の根本的な改善には至らない
根治的アプローチとしての肝移植:画期的な成果と課題
食事療法の明確な限界を乗り越える手段として、近年肝移植(Liver Transplantation)が注目されています。肝臓は体内のメチオニン代謝の主要な場であるため、正常なSAHH酵素活性を持つ健康な肝臓を移植することで、全身の代謝ホメオスタシスが劇的に改善するという仮説に基づいています。
ピッツバーグ小児病院で生後40ヶ月に肝移植を受けた男児の症例では、肝臓の血流再開からわずか24時間以内にメチオニン・SAMが完全に正常化し、SAHが96%減少、SAM/SAH比が即座に倍増して正常範囲に回復しました。術後6ヶ月の追跡で食事制限なしの通常タンパク質摂取下でもこれらの正常状態を完璧に維持しました。
7. 遺伝カウンセリング
AHCY欠損症の確定診断後には、患者本人と家族への丁寧な遺伝カウンセリングが不可欠です。遺伝カウンセリングは確率の押し付けではなく、家族が主体的に意思決定できるよう情報提供と心理的サポートを行うものです。
- ➤再発リスクの説明:常染色体劣性遺伝のため、両親がともにキャリアの場合、次の妊娠における発症確率は理論上25%です。
- ➤きょうだいへの保因者検査:患者のきょうだいも50%の確率でキャリアである可能性があります。将来の家族計画のために拡張型保因者スクリーニング(女性)や男性向け保因者スクリーニングの受検を検討できます。
- ➤次子への出生前診断:既知の変異が同定されていれば、次の妊娠時に絨毛検査・羊水検査による出生前遺伝子診断が可能です。妊娠前の相談については妊娠前遺伝子検査をご参照ください。
- ➤キャリアスクリーニングの意義:本疾患のような常染色体劣性疾患に対するキャリアスクリーニングの意義については、ACMG・ACOGのキャリアスクリーニング推奨内容もご参照ください。
- ➤心理的サポート:遺伝性疾患のキャリアとして検査を受けた方の体験については、遺伝性疾患と家族計画:あきらめないための選択肢もあわせてご覧ください。
💡 用語解説:保因者(キャリア)とは
常染色体劣性遺伝疾患において、2本の染色体のうち1本だけに変異を持つ人を「保因者(キャリア)」と呼びます。保因者自身は通常発症しませんが、パートナーも同じ遺伝子の保因者だった場合、お子さんが発症する可能性があります。AHCY欠損症のような超希少疾患の保因者は、何世代もの間気づかれないまま受け継がれていることがほとんどです。
8. よくある誤解
誤解①「メチオニン正常ならAHCY欠損症ではない」
報告症例の約半数で初期メチオニン値が正常です。p.G71S変異の症例では生後8ヶ月まで完全に正常でした。メチオニン正常はAHCY欠損症を除外する根拠になりません。
誤解②「SAMeサプリは代謝を助けるから有益では?」
SAMe(市販サプリ)はAHCY欠損症では絶対禁忌です。すでにSAMが病的蓄積しているため、SAMeの追加摂取は病態を著しく悪化させる危険があります。一般向けサプリ情報のまま摂取しないでください。
誤解③「食事療法で完全に治る疾患」
メチオニン制限食により代謝マーカーはある程度改善しますが、SAM・SAHは正常化せず、DNAの異常な高メチル化も34ヶ月の治療後も残存します。精神運動発達が年齢相応に追いつくことはありません。
誤解④「初期症状がなければ安心」
初期の症状や所見の欠如は決してAHCY欠損症を除外する根拠になりません。26歳まで診断されなかった例もあります。強い臨床的疑いがあれば積極的にSAM・SAH測定と遺伝子解析を検討すべきです。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 AHCY欠損症・先天代謝異常の診断・遺伝カウンセリングについて
超希少遺伝性疾患のご相談、保因者検査、出生前診断に関するお問い合わせは
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にどうぞ。
関連記事
参考文献
- [1] Orphanet. S-adenosylhomocysteine hydrolase deficiency. ORPHA:88618. [Orphanet]
- [2] NIH GARD. Hypermethioninemia with deficiency of S-adenosylhomocysteine hydrolase. [NIH GARD]
- [3] Bukovska G, et al. S-adenosylhomocysteine hydrolase deficiency in a human: A genetic disorder of methionine metabolism. Proc Natl Acad Sci USA. 2004;101(12):4234-4239. [PNAS]
- [4] Stender S, et al. The biochemical profile and dietary management in S-adenosylhomocysteine hydrolase deficiency. Mol Genet Metab. 2022;137(1-2):118-127. [PMC9249945]
- [5] Baric I, et al. S-Adenosylhomocysteine Hydrolase (AHCY) Deficiency: Two Novel Mutations with Lethal Outcome. PLoS Genet. 2005;1(1):e13. [PMC2876820]
- [6] Grubbs R, et al. S-Adenosylhomocysteine hydrolase deficiency: A second patient, the younger brother of the index patient, and outcomes during therapy. Mol Genet Metab. 2010;100(1):62-72. [PMC2441944]
- [7] Honzik T, et al. Adult-onset liver disease and hepatocellular carcinoma in S-adenosylhomocysteine hydrolase deficiency. Mol Genet Metab. 2016;117(2):111-117. [PMC4733618]
- [8] Ahlin A, et al. Common pattern of coagulation abnormalities in S-adenosylhomocysteine hydrolase deficient patients. JIMD Rep. 2020;54(1):41-48. [ResearchGate]
- [9] Mazariegos GV, et al. Liver transplantation for treatment of severe S-adenosylhomocysteine hydrolase deficiency. Pediatr Transplant. 2015;19(5):531-535. [ResearchGate]
- [10] Bjornsson HT, et al. Abnormal Hypermethylation at Imprinting Control Regions in Patients with S-Adenosylhomocysteine Hydrolase (AHCY) Deficiency. PLoS One. 2016;11(3):e0151261. [PMC4790936]
- [11] Yin H, et al. Functional and Pathological Roles of AHCY. Int J Mol Sci. 2021;22(5):2575. [PMC8044520]
- [12] Frikke-Schmidt H, et al. Dysmorphic Findings in SAHH Deficiency with a Novel Variant in the AHCY Gene. Mol Syndromol. 2024;15(6):531-537. [PMC11614430]
- [13] MedlinePlus Genetics. AHCY gene. National Library of Medicine. [MedlinePlus]






