目次
- 1 1. aEDS関節弛緩型1とは何か:疾患の概要と疫学
- 2 2. 原因遺伝子COL1A1と遺伝形式
- 3 3. 分子病態メカニズム:エクソン6スキッピングが引き起こすもの
- 4 4. COL1A1疾患スペクトラム:骨形成不全症(OI)との重複
- 5 5. 多系統にわたる臨床症状
- 6 6. 診断基準:2017年国際分類と確定診断
- 7 7. 鑑別診断:他のEDS病型・類縁疾患との見分け方
- 8 8. 出生前検査・遺伝学的検査:COL1A1を調べる
- 9 9. 治療・管理:多職種チームによる対症療法
- 10 10. 長期予後と生活支援・合理的配慮
- 11 11. 最新研究動向と将来の展望(2024–2026年)
- 12 よくある質問(FAQ)
- 13 関連記事
- 14 参考文献

📍 クイックナビゲーション
エーラス・ダンロス症候群関節弛緩型1(aEDS type 1)は、出生時から両側の股関節が脱臼していることを最大の特徴とする超希少な遺伝性結合組織疾患です。原因はCOL1A1遺伝子の変異で、世界で約42例しか報告されていないほど稀ですが、生涯にわたる運動機能と生活の質に深刻な影響を及ぼします。骨形成不全症(OI)との重複という重要な側面も近年明らかになっています。
Q. aEDS関節弛緩型1(COL1A1)とはどのような病気ですか?
A. COL1A1遺伝子のエクソン6スキッピングによりI型コラーゲンの形成が障害される、常染色体顕性遺伝の超希少疾患です。出生時の両側股関節脱臼が最大の特徴で、全身性の著しい関節弛緩・皮膚の過伸展性・筋緊張低下を伴います。同じCOL1A1が原因の骨形成不全症と表現型が重なり、骨折リスクの亢進を伴う点が、より軽症のCOL1A2型(type 2)との違いです。根治療法はなく、多職種連携による長期的な対症療法が中心となります。
- ➤原因 → COL1A1遺伝子のエクソン6スキッピングによるI型コラーゲン形成不全(type 1)
- ➤頻度 → 100万人あたり1人未満の超希少疾患。世界報告症例は約42例
- ➤症状 → 出生時両側股関節脱臼・全身性関節弛緩・筋緊張低下・皮膚過伸展性・骨折リスク亢進
- ➤診断 → 2017年国際臨床診断基準+COL1A1遺伝子検査・生化学的コラーゲン解析による確定
- ➤出生前検査 → COL1A1はNIPT(インペリアルプラン)でも調べられるようになりました
1. aEDS関節弛緩型1とは何か:疾患の概要と疫学
エーラス・ダンロス症候群(EDS)は、コラーゲンをはじめとする結合組織の生合成・構造・修飾プロセスに関わる遺伝的欠陥によって生じる、臨床的・遺伝的に多様な遺伝性疾患群です。2017年の国際EDSコンソーシアムによる再分類では、臨床的特徴と分子遺伝学的基盤に基づいて13の独立した病型に分けられています。
その中でも関節弛緩型EDS(Arthrochalasia EDS:aEDS)は、最も稀有でありながら極めて重篤な身体障害をもたらす病型の一つです。歴史的には「EDS VII型」と呼ばれ、原因遺伝子によってさらに2つの型に細分されます。本記事で扱うaEDS 1型(旧EDS VIIA型)はCOL1A1遺伝子の変異によるもので、COL1A2変異による2型(旧EDS VIIB型)とは別の病型として扱われます(type 2については別記事で解説します)。
有病率:aEDSは100万人あたり1人未満とされる「超希少疾患(ultra-rare disorder)」です。全世界の医学文献で報告されている症例数は約42例にとどまります。EDS全体の有病率が3,100〜5,000人に1人であることと比較すると、その稀少さが際立ちます。
結合組織とは、骨・皮膚・腱・靭帯・血管壁など、体のあらゆる部位で「構造的な足場」を担う組織の総称です。その主要な構成タンパク質がコラーゲンです。結合組織が正常に機能しないと、関節・皮膚・骨など多系統にわたる症状が現れます。aEDS 1型ではこのコラーゲン(特にI型コラーゲン)の形成そのものが遺伝子レベルで障害されています。
日本国内では、EDS全体が指定難病168として認定されており、医療費助成の対象となっています。aEDSはICD-10コード「Q79.6(エーラス・ダンロス症候群)」に分類され、稀少かつ重篤な病型として高度な専門的介入が必要な疾患に位置づけられています。
2. 原因遺伝子COL1A1と遺伝形式
aEDS 1型は常染色体顕性遺伝(常染色体優性遺伝)の形式をとります。つまり、両親のいずれかから変異を持つアレルを1つ受け継ぐだけで発症します。患者が子どもをもうける場合、その変異が次世代に伝わる確率は各妊娠において50%です。また、家族歴がない孤発例(de novo変異)として生じるケースも報告されています。
23対ある染色体のうち、性染色体(X・Y染色体)以外の「常染色体」上の遺伝子に変異があり、対になる遺伝子の片方(1つのアレル)だけに変異があっても発症する遺伝形式です。「顕性(優性)」とは「1コピーで効果が現れる」という意味であり、「強い・弱い」の意味ではありません。両親のどちらかが患者であれば、子への遺伝確率は50%です。
原因遺伝子であるCOL1A1(第17番染色体長腕17q21.33)は、I型コラーゲンを構成するプロα1(I)鎖をコードしています。正常なI型コラーゲンは、2本のプロα1(I)鎖と1本のプロα2(I)鎖が結合した三量体(トリマー)です。
▍COL1A1型(type 1)が重症化しやすい理由
I型コラーゲンはα1鎖2本+α2鎖1本で構成されます。α1鎖をコードするCOL1A1に変異があると、確率的に三量体全体に異常鎖が取り込まれる割合が高くなり、後述するドミナントネガティブ効果がより強く現れます。このため、COL1A1によるaEDS 1型は、COL1A2によるaEDS 2型と比べて筋骨格系の症状がより重篤になる傾向があると複数の研究で指摘されています。さらに1型では、骨形成不全症と同様の骨減少症・骨折リスクの亢進を伴う点が特徴です(詳細はセクション4)。
変異の浸透率(penetrance)は男女ともにほぼ100%と推定されており、変異を持っていれば必ず何らかの臨床症状が現れます。ただし、重症度には個体差や家族間でのばらつきが存在します。
🔗 あわせて読む:COL1A1遺伝子の役割や、この遺伝子が含まれる検査プランの詳細はCOL1A1遺伝子ページで解説しています。
3. 分子病態メカニズム:エクソン6スキッピングが引き起こすもの
aEDS 1型の根底にある分子異常は、単なるタンパク質量の低下ではなく、RNAのスプライシング過程における特異的な欠失(エクソン6スキッピング)と、それに続くコラーゲン分子の立体的な構造異常です。
DNAからタンパク質が作られる過程では、まず「プレmRNA」が作られ、その後「スプライシング」という編集作業でイントロン(不要部分)が取り除かれ、エクソン(必要部分)だけがつなぎ合わされます。「エクソン6スキッピング」とは、スプライス部位の変異によりエクソン6が誤って丸ごと除去されてしまう現象です。COL1A1のエクソン6には、プロコラーゲンの切断に必要な酵素切断部位と、コラーゲン分子同士を架橋するために必須のリジン残基が含まれています。
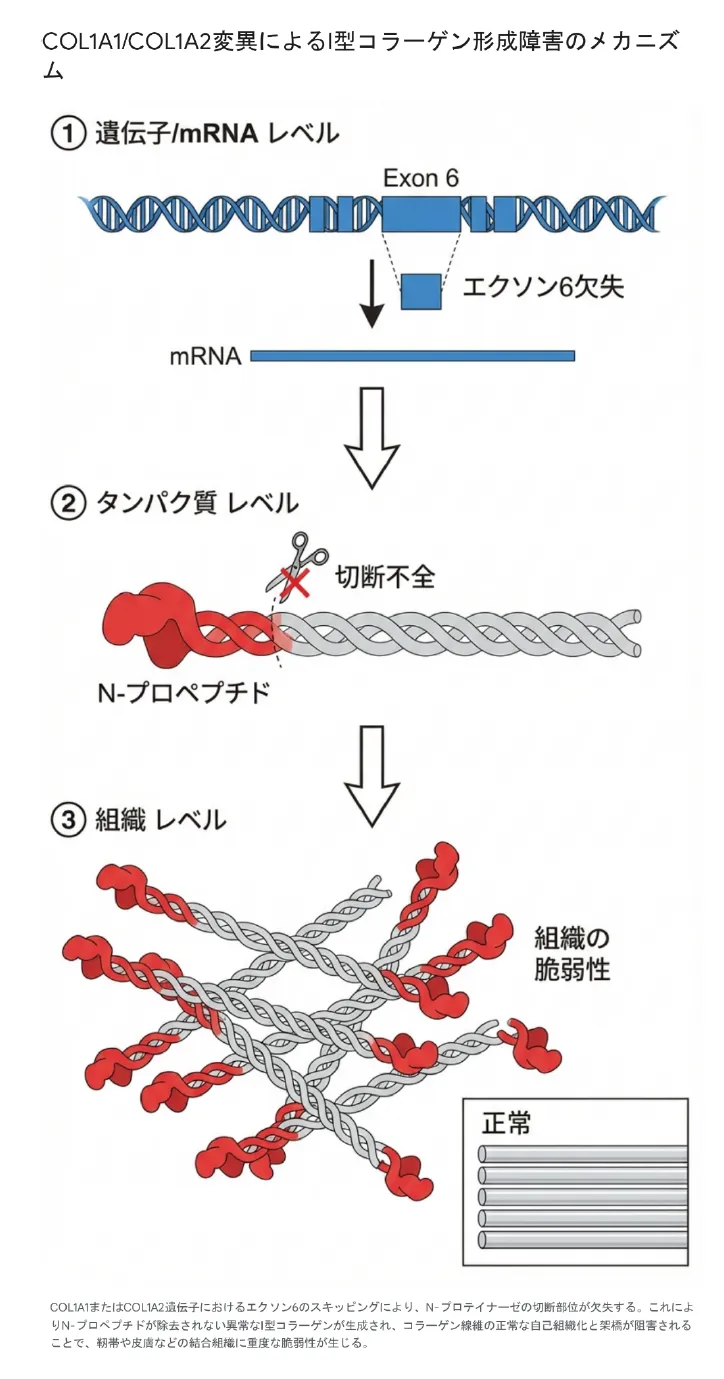
正常なI型コラーゲンが完成するには、前駆体であるプロコラーゲンのN末端(アミノ末端)にある「N-プロペプチド」を、ADAMTS-2などの酵素(N-プロテイナーゼ)が特異的に切断する必要があります。エクソン6がスキッピングされると、この酵素の切断部位がコードされなくなり、N-プロペプチドが残ったままの異常なコラーゲン分子(pN-コラーゲン)が細胞外に分泌・蓄積されます。
この切り離されなかった巨大なN-プロペプチドが立体障害(steric hindrance)を引き起こし、コラーゲン分子間の規則正しい架橋形成と平行配列を著しく阻害します。電子顕微鏡下では、形成された細線維は正常より細く・直径が不均一・密度が低い不規則な構造を示します。その結果、皮膚・靭帯・関節包・骨などにおけるコラーゲン線維の強度が根本的に失われ、aEDSの臨床症状として表れます。
変異によって生じた異常タンパク質が、正常なタンパク質とも結合して三量体(3本のらせん構造)を形成し、正常な機能まで阻害してしまう現象です。aEDS患者の線維芽細胞は、正常なプロα1(I)鎖と短縮型の異常鎖をほぼ同量ずつ合成しますが、異常鎖が正常分子に取り込まれることで、ハプロ不全(タンパク質が半減するだけ)よりもはるかに深刻な結合組織の破壊をもたらします。これがaEDSが骨形成不全症の軽症型と異なり、多系統に重篤な症状を示す分子生化学的な理由です。
4. COL1A1疾患スペクトラム:骨形成不全症(OI)との重複
COL1A1は、aEDSだけでなく骨形成不全症(Osteogenesis Imperfecta:OI)の主要な原因遺伝子でもあります。同じ遺伝子から、変異の種類によって異なる疾患が生じるため、aEDS 1型を理解するうえでこの「疾患スペクトラム」の視点が欠かせません。
一般に、OIの最も軽度なタイプ(OI 1型)はCOL1A1のハプロ不全(正常なコラーゲンが半減)で起こるのに対し、重症型のOIやaEDSはコラーゲン分子の構造を破壊する変異(ドミナントネガティブ)で起こります。つまりaEDS 1型は、分子病態のうえでもOIと地続きの位置にあります。
▍aEDS 1型で見逃せない「骨折リスク」
骨折の多発はaEDS全般の典型像ではないとされてきましたが、COL1A1変異によるaEDS 1型では、放射線学的な骨減少症やウォーム骨(縫合間骨)が高頻度に認められ、骨折への脆弱性が有意に亢進することが報告されています。Ayoubらによる12例のコホート研究では9例に骨折歴が認められました。出産時の外傷による病的頭蓋骨骨折、長管骨・鎖骨・手指の骨折、軽微な衝撃での脊椎圧迫骨折などの症例があり、骨脆弱性は本型における重要な合併症です。
近年、EDSに特徴的な「重度の関節弛緩や皮膚症状」と、OIに特徴的な「短身長・青色強膜・骨折しやすさ」の両方を併せ持つ患者群が存在することがわかってきました。これらは従来の基準ではaEDSにもOIにも完全には合致しないため、Morlinoらにより「COL1関連オーバーラップ症候群」という疾患概念が提唱されています。2017年の国際分類では公式な独立疾患としてはまだ認定されていませんが、aEDS 1型の一部はこのスペクトラム上に位置づけられ、単なる関節疾患ではなく潜在的な骨脆弱性を伴う全身性結合組織疾患として管理することが求められます。
同じCOL1A1から生じる代表的な疾患を比較すると、以下のようになります。
| 疾患名 | COL1A1変異の型 | 重症度 | 主要な特徴 |
|---|---|---|---|
| aEDS 1型(旧EDS VIIA) | エクソン6欠失 | 中等度〜重度 | 先天性両側股関節脱臼、重度の関節弛緩、皮膚過伸展、筋緊張低下、骨減少症・骨折リスク亢進 |
| 骨形成不全症1型(OI type 1) | ハプロ不全 | 軽度 | 青色強膜、骨折しやすいが変形は伴わない、ほぼ正常な身長 |
| 骨形成不全症3型(OI type 3) | 構造変異 | 重度(進行性) | 重度の短身長、頻回な骨折と長管骨変形、象牙質形成不全、脊柱変形 |
| カフェイ病(Caffey病) | Arg836Cys変異 | 軽度〜重度 | 乳児期発症の皮質骨肥厚(過骨症)、軟部組織腫脹、発熱と疼痛。通常2歳までに自然軽快 |
5. 多系統にわたる臨床症状
aEDS 1型の患者は出生直後から成人まで、多くの臓器・組織にわたる症状を呈します。I型コラーゲンが体のあらゆる部位に存在するため、症状は筋骨格系だけにとどまりません。
① 筋骨格系・整形外科的異常
先天性両側股関節脱臼
aEDSの決定的ホールマーク(指標)です。ほぼ全例が出生時にこの所見を持ちます。胎児期からの関節包の異常弛緩と寛骨臼(ソケット)の浅い形成が複合して生じます。この有無が診断の出発点となります。
重度の全身性関節過可動性
肩・膝・肘などの大関節から、手指・足趾・手首などの小関節まで、正常な可動域をはるかに超えた過可動性があります。乳児期には日常のおむつ替えや着替えでさえ脱臼を来すほど関節が不安定です。
反復性脱臼・変形性関節症
四肢のあらゆる関節で日常的な反復性脱臼・亜脱臼が生じます。慢性的な微小外傷が蓄積し、関節軟骨の早期摩耗から若年性変形性関節症を招きます。
重度の脊椎変形
小児期早期から後側彎症(後彎+側彎の合併)が進行します。重度の後側彎症は胸腔を圧迫し、胸郭不全症候群による致死的な呼吸機能障害をもたらすリスクがあります。
その他、先天性の内反尖足・重度の扁平足・外反膝なども高頻度で見られます。前述のとおり、放射線学的には軽度の骨減少症や、頭蓋骨縫合線に沿ったウォーム骨(縫合間骨)が観察されることもあります。
② 神経・筋機能への影響
出生時からの重度の筋緊張低下(ヒポトニア)がほぼ例外なく認められます。筋肉が「フロッピー(ぐにゃぐにゃ)」な状態となり、首のすわり・寝返り・座位・歩行などの運動発達マイルストーンが著しく遅れます。重症例では出生直後に呼吸筋の脆弱性から低換気をきたし、nCPAP(経鼻持続的気道陽圧)などの呼吸補助を要することもあります。この極端な筋力低下から、乳幼児期には脊髄性筋萎縮症(SMA)などの神経筋疾患と誤診されるリスクがあります。
重要ポイント:aEDSのヒポトニアは神経・筋疾患によるものではなく、結合組織の欠陥に由来する二次的なものです。年齢を重ねるにつれて徐々に改善する傾向があり、SMAなどとは異なります。神経学的反射は保たれており、関節の過可動性・皮膚の特徴との組み合わせで鑑別します。また、aEDSは知能・認知の発達には直接の悪影響を及ぼさず、知的発達は正常に進みます。
また、頸椎の不安定性からキアリ奇形(小脳扁桃の下垂)が合併することがあります。硬膜のコラーゲン異常による脳脊髄液(CSF)漏出は頭蓋内圧低下を引き起こし、体位性頭痛・羞明・複視などの症状の原因となります。
③ 皮膚・結合組織の異常
皮膚は異常に伸びやすく、引っ張ると正常範囲を大きく超えて伸展しますが、放すと即座に元の位置に戻ります(たるんだ余剰皮膚=皮膚弛緩症とは異なります)。触感は非常に柔らかく、ビロードのようと表現されます。手掌・足底に細かいシワ(クリスクロス・パターン)が見られることもあります。
軽微な外傷で容易に挫傷(内出血・血腫)が生じ、創傷治癒が遅延します。治癒後には陥凹したシワ状の萎縮性瘢痕(cigarette-paper scars)が形成されます。ただし皮膚症状の重症度は、古典型EDS(cEDS)と比べて一般的に軽度です。この皮膚・皮下組織の脆弱性は、後述する整形外科治療でのギプス固定・装具装着の際に深刻な皮膚トラブル(褥瘡・壊死)を招く最大の要因となります。
④ 頭蓋顔面・心血管の特徴
小児期には、小顎症・両眼開離(目の間隔が広い)・内眼角贅皮・前頭部突出・大きな泉門の開存などの特徴的な顔貌が見られます。これらの顔貌的特徴はヒポトニアと同様、年齢とともに目立たなくなる傾向があります。血管型EDSのような致死的な大血管・消化管の自発破裂は典型的ではありませんが、一部の患者で僧帽弁・大動脈弁・三尖弁の弁膜症が報告されており、心血管系のベースライン評価が推奨されます。
6. 診断基準:2017年国際分類と確定診断
aEDSの診断は、①臨床的評価と②生化学的・分子遺伝学的検査の二段階で行われます。2017年の国際EDSコンソーシアムにより、aEDSの大基準・小基準が明確に定義され、診断の標準化が実現しました。
▍大基準(Major Criteria)
- 大基準1(必須)先天性両側股関節脱臼 ── 出生時のX線または超音波検査で確認。aEDS診断のゲートウェイ
- 大基準2重度の全身性関節過可動性(複数関節での脱臼または亜脱臼を伴う)
- 大基準3皮膚の過伸展性(引っ張ると正常範囲を超えて伸び、放すと速やかに戻る)
▍小基準(Minor Criteria)
- ●筋緊張低下(ヒポトニア)
- ●後側彎症(小児期早期から進行する後彎と側彎の合併)
- ●軽度の骨減少症(放射線学的に確認される骨密度低下)
- ●組織の脆弱性(萎縮性瘢痕形成を含む)
- ●挫傷を生じやすい皮膚(軽微な接触による内出血・血腫)
遺伝学的検査への適応条件:「大基準1+大基準3」を満たすか、「大基準1+大基準2+小基準2項目以上」を満たす場合。いずれのパスでも大基準1(先天性両側股関節脱臼)が必須要件です。
臨床基準を満たした場合、確定診断のためには検査室レベルの解析が行われます。
分子遺伝学的検査
血液・唾液などからDNAを抽出し、次世代シーケンサー(NGS)やサンガー法でCOL1A1のエクソン6スキッピングを引き起こす病原性バリアント(主にスプライス部位変異)を同定します。これが確定診断の必須要件です。
生化学的コラーゲン解析
皮膚生検から培養した線維芽細胞のI型コラーゲンを電気泳動で解析し、N末端プロペプチドが切断されない「pN-α1(I)鎖」のバンド(ダブレット)を検出します。これがaEDS 1型の生化学的な確定証拠となります。
関節過可動性を数値化する国際的な評価指標です。小指の背屈・親指の手首への接触・肘の過伸展・膝の過伸展・立位前屈時の手掌の床接触という9点満点のスコアで、5点以上がハイパーモビリティのカットオフ値です。aEDSでは全身性の高度な関節弛緩のため通常は高値(多くは満点近く)を示します。ただし乳幼児期は基準値の適用に注意が必要です。
7. 鑑別診断:他のEDS病型・類縁疾患との見分け方
aEDSの症状は他のEDS病型や結合組織疾患と部分的に重複するため、精緻な鑑別が不可欠です。なお、同じCOL1A1から生じる骨形成不全症・カフェイ病との関係はセクション4を参照してください。
| 疾患名 | 関連遺伝子 | 遺伝形式 | aEDS 1型との決定的な相違点 |
|---|---|---|---|
| aEDS 2型 | COL1A2 | 常染色体顕性 | 同じaEDSだが、筋骨格症状が1型より軽度の傾向。骨折リスクの亢進は1型でより顕著 |
| 古典型EDS (cEDS) | COL5A1, COL5A2 | 常染色体顕性 | 非常に脆弱な皮膚と重度の萎縮性瘢痕が顕著。先天性股関節脱臼は必須ではない |
| 血管型EDS (vEDS) | COL3A1 | 常染色体顕性 | 大動脈解離・腸管破裂のリスクが極めて高く致死的。皮膚の過伸展性は乏しい |
| 後側彎型EDS (kEDS) | PLOD1, FKBP14 | 常染色体潜性 | 後側彎症・筋緊張低下は共通するが、強膜破裂・難聴を伴い潜性遺伝である点が異なる |
| 皮膚脆弱型EDS (dEDS) | ADAMTS2 | 常染色体潜性 | N-プロテイナーゼ酵素自体の欠損。過剰なたるんだ皮膚と重度の皮膚裂傷が特徴。股関節脱臼は主徴ではない |
| ラーセン症候群 | FLNB | 顕性/潜性 | 多発性関節脱臼を伴うが、特有の平坦な顔貌と頸椎の異常が際立つ。コラーゲン異常による皮膚の過伸展性は見られない |

8. 出生前検査・遺伝学的検査:COL1A1を調べる
aEDS 1型の原因であるCOL1A1は、出生後の遺伝学的検査だけでなく、出生前のNIPT(無侵襲的出生前遺伝学的検査)でも調べられるようになりました。
ミネルバクリニックのインペリアルプランでは、母体採血のみ(非侵襲)で胎児由来のDNAを解析する単一遺伝子NIPTの対象遺伝子にCOL1A1が含まれています(陽性的中率>99.9%)。aEDS 1型や、同じCOL1A1が原因の骨形成不全症が気がかりなご家族にとって、選択肢の一つとなります。
臨床的な位置づけ:aEDSはde novo(新生)変異や顕性遺伝で生じるため、NIPTは家系内に既知のCOL1A1バリアントがある場合や、de novoの顕性疾患スクリーニングとして意義があります。NIPTは確定診断ではないため、陽性の場合は羊水検査・絨毛検査による確定診断が次のステップとなります。
なお、COL1A1の検査はNIPT(出生前)だけに限りません。出生後・診断目的の通常の遺伝学的検査でも調べることができます。どの検査が適しているかは、ご家族の状況(家族歴の有無、検査の目的、妊娠中か否か)によって異なるため、遺伝カウンセリングで一緒に整理していくことをおすすめします。
🧬 COL1A1遺伝子ページ遺伝子の機能や、含まれる検査プランの一覧を解説。
🔍 羊水・絨毛検査NIPT陽性後の確定診断について解説。
9. 治療・管理:多職種チームによる対症療法
aEDS 1型に対する根治療法は現在のところ存在せず、治療はすべて対症療法です。患者の身体的安定性・運動機能・生活の質の最大化を目指し、整形外科医・理学療法士・臨床遺伝専門医・皮膚科医・疼痛管理専門医・心理士などによる学際的チームアプローチが不可欠です。
① 股関節脱臼への外科的介入と「治療法別成功率」
通常の発達性股関節形成不全(DDH)に用いるパブリック・ハーネスやギプスなどの保存的整復術はaEDS患者にはほとんど効果がなく、安定した整復の成功例は皆無です。脆弱な皮膚への持続的圧迫で重度の皮膚潰瘍・壊死を招くうえ、長期固定が筋萎縮を悪化させ、固定解除後に即再脱臼するためです。関節包のみを縫縮する軟部組織手術も再脱臼率が極めて高いとされます。
aEDSにおける先天性股関節脱臼の治療法別 安定整復成功率
(ギプス等)観血的整復術
(縫縮のみ・1/10)観血的整復術
(骨切り術併用・2/6)
過去の文献で報告された安定整復の達成率。保存的治療は全例で失敗。外科的介入では、腸骨骨切り術(必要に応じ大腿骨骨切り術を併用)を行った場合のみ部分的な成功(約33%)が得られているが、全体として成功率は極めて低い。
医学文献が示す相対的に有効な手段は、腸骨骨切り術(Salter法・Pemberton法など)で骨格の形状そのものを改変し、寛骨臼を深くして弛緩した関節包に依存せず大腿骨頭を保持する方法です。それでも成功率は約33%にとどまります。このため現代のマネジメントでは、自立歩行の獲得見込みが極めて低いと判断される場合、不必要な侵襲的手術を意図的に「保留(Deferral)」し、内反足の矯正など日常生活・移動手段の確保に治療の焦点を移すことが、患者の総合的な利益にかなうと考えられています。
人工股関節置換術(THA)のリスク:EDS患者はTHA後の不安定性・再脱臼リスクが対照群の約3.2倍、インプラントの無菌的ゆるみのリスクは約3.8倍、全原因による再手術リスクは約2.4倍に達するとの研究データがあります。手術適応は極めて慎重に判断する必要があります。
② 脊椎変形の外科的管理:MAGECロッドという革新
早期発症型の重度後側彎症は、放置すれば胸郭不全症候群(肺の成長阻害による呼吸不全)を招くため矯正・固定が必要です。しかし従来の成長ロッドは半年〜1年ごとの皮膚切開が必要で、消毒のワイプで皮膚が裂けるほど脆弱なaEDS患者では創傷治癒不全・重症感染症のリスクが極めて高く、実施が困難でした。
MAGnetic Expansion Controlロッドの略称です。一度体内に埋め込んだ後、体外から磁力を当てるだけで非侵襲的にロッドを伸長できる脊椎固定デバイスです。繰り返しの切開手術を回避できるため、皮膚が極度に脆弱なaEDS患者で重度の脊椎変形を17年間にわたり矯正に成功した長期追跡症例が報告されており、aEDSの脊椎管理における新たなパラダイムとして評価されています。
③ 骨密度の管理と理学療法
COL1A1型では骨形成不全症との重複として骨折リスクが高いため、定期的なDXAスキャン(骨密度測定)とビタミンD・カルシウムの管理が基本となります。骨折を反復する症例や著しい骨密度低下例には、骨形成不全症の治療に準じてビスホスホネート製剤の投与が検討され、骨の強化と骨折予防に一定の有用性が報告されています。
理学療法は管理戦略の中核です。基本方針は「start low and go slow」(関節への負荷を極めて少なくした等尺性エクササイズから始め、ゆっくり進む)です。
- ➤固有受容感覚の再訓練:関節の位置感覚が鈍麻しているため、鏡を使ったフィードバックやバランス訓練でニュートラルな関節位置の保持を学習させます
- ➤装具(KAFO・AFO等)の活用:皮膚が極度に脆弱なため、装具のフィッティング調整と毎日の皮膚状態の監視がセットで必要です
- ➤避けるべき運動:ランニング・ジャンプ・コンタクトスポーツ(ラグビー・柔道等)・関節の過伸展を強調するヨガや体操は脱臼と軟骨損傷を誘発するため厳格に避けます
- ➤水中運動(アクアセラピー):関節への衝撃が少なく、aEDS患者に適した運動形態として推奨されることがあります
④ 心血管モニタリング・創傷管理・疼痛マネジメント
大動脈基部拡張や弁膜症の有無を評価するベースラインの心エコー検査をすべての患者に実施します。創傷縫合の際は通常より深く縫合糸をかけ、抜糸まで通常の2倍程度の期間を確保し、Steri-Stripsや組織接着剤の併用が推奨されます。慢性疼痛にはNSAIDs・アセトアミノフェンを基本とし、難治例ではペインクリニックでの脊髄刺激療法等も慎重に検討されます。幼少期からの絶え間ない痛みと身体的制限がもたらす精神的ストレスは大きく、認知行動療法(CBT)などの心理的サポートも不可欠です。
10. 長期予後と生活支援・合理的配慮
最も特筆すべきは、aEDSの生命予後(寿命)は一般集団と比べて劣るものではないという点です。血管型EDSのような致死的な大血管・臓器破裂を主徴としないためです。一方で、身体機能とQOLの観点では道のりは過酷です。日常的な関節脱臼が関節軟骨の摩耗を招き、若年期からの早期変形性関節症が進行します。
Ayoubらの12例コホート追跡では、4例(約33%)が最終的に自力歩行が不可能となり車椅子に依存していました。特にCOL1A1変異の1型は筋骨格系合併症がより重症化しやすく、より集中的・長期的なサポートが必要です。乳幼児期に顕著だった筋緊張低下・関節過可動性は加齢とともにある程度改善し、適切な介入で小児期後期に自力歩行が可能になる例も報告されますが、関節の根本的な不安定性は成人後も残存し、骨減少症・骨折リスクも生涯継続するため、成人以降も1〜2年ごとのDXAスキャンが推奨されます。
重要なのは、aEDSは知能・認知の発達に直接の悪影響を及ぼさないことです。適切なペインマネジメント、バリアフリー化、モビリティ支援、そして教育・職場での社会的支援が整えば、学業を全うし、キャリアを築き、社会的に自立した充実した人生を送ることは十分に可能です。
教育現場・職場における合理的配慮
物理的・環境的配慮
エルゴノミクスチェアの導入、定期的な横臥休息時間の確保、重いカバンの免除、教科書のデジタル化、エレベーター利用、車椅子使用の許可など
学習・書字への配慮
試験時間の延長、音声入力ソフトやPCの使用許可、太いグリップの筆記具の提供、代筆者の配置など
視覚・体育への配慮
教室照明の調整、文字の拡大・コントラスト強調、体育は医師・理学療法士の指導に基づく適応型プログラムへの変更
超希少疾患を抱えながら生きることは、身体的苦痛だけでなく、周囲の無理解による孤立感や不確実な未来への不安など深刻な精神的負担を伴います。心理的ケアや患者支援団体を通じたピアサポートへのアクセスが強く推奨されます。
11. 最新研究動向と将来の展望(2024–2026年)
「The Road to 2026」イニシアチブ
国際EDSコンソーシアムが2017年基準のアップデートを推進中です。2024〜2025年に実施された世界規模の患者調査の知見を反映し、2026年後半に新たな分類基準と管理ガイドラインの発表が予定されています。aEDSを含む希少病型の診断アルゴリズムがさらに洗練される見込みです。
EDS/HSDグローバル・バイオバンク
The Ehlers-Danlos Societyが250万ドルの寄付を受け、2025年にEDS/HSDのグローバル・バイオバンクを立ち上げ。EDS/HSDの当事者と非当事者から生体サンプルを収集し、遺伝子・タンパク質・組織レベルの研究を支援します。aEDSのような超希少型の理解と個別化医療の基盤整備も加速すると期待されます。
分子標的治療の研究
COL1A1のエクソン6スキッピングをインビトロのミニ遺伝子アッセイで再現・解析する手法が確立されています。将来的にはアンチセンス・オリゴヌクレオチド(ASO)などのRNAモジュレーション技術で異常なスプライシングを補正する根本的な分子標的薬の開発が期待されています。
よくある質問(FAQ)
関連記事
参考文献
- [1] Malfait F, et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017. [PubMed]
- [2] The Ehlers-Danlos Society. aEDS(関節弛緩型EDS)疾患情報. [Ehlers-Danlos Society]
- [3] Sobey G, et al. Ehlers–Danlos Syndrome Type Arthrochalasia: A Systematic Review. PMC 2022. [PMC]
- [4] Ayoub S, et al. Clinical features, molecular results, and management of 12 individuals with arthrochalasia EDS. 2020. [PubMed]
- [5] Giunta C, et al. Ehlers-Danlos Arthrochalasia type (VIIA-B) – expanding the phenotype. PMC 2014. [PMC]
- [6] Orphanet. Arthrochalasia Ehlers-Danlos syndrome. [Orphanet]
- [7] Geneskin. Ehlers-Danlos Syndrome, Arthrochalasis Type(専門医向け情報). [Geneskin]
- [8] F1000Research. Essential timing of orthopaedic treatment in arthrochalasia EDS. [F1000Research]
- [9] Morlino S, et al. COL1-related overlap disorder. The Ehlers-Danlos Society. [EDS Society]
- [10] Baban A, et al. Seventeen-year outcome of surgical management of severe early onset kyphoscoliosis in arthrochalasia-type EDS. PMC 2025. [PMC]
- [11] COL1A1- and COL1A2-Related Osteogenesis Imperfecta. GeneReviews, NCBI. [GeneReviews]
- [12] The Ehlers-Danlos Society. The Road to 2026 / Global Biobank. [EDS Society]
- [13] 厚生労働省難病情報センター. エーラス・ダンロス症候群(指定難病168). [難病情報センター]






