OCA2遺伝子
承認済シンボル:OCA2
遺伝子名:OCA2 melanosomal transmembrane protein
参照:
HGNC: 8101
NCBI:4948
遺伝子OMIM番号611409
Ensembl :ENSG00000104044
UCSC :
AllianceGenome : HGNC : 8101
遺伝子のlocus type :タンパク質をコードする
遺伝子のグループ:
遺伝子座: 15q12-q13.1
OCA2遺伝子の機能
OCA2遺伝子産物は、細胞内クロライドチャネル活性を可能にする。リソソーム内腔のpH上昇およびチロシンからのメラニン生合成過程に関与。オルガネラの結合膜および小胞体膜に存在。光線性角化症、眼皮膚アルビニズムII型、色素沈着症、皮膚癌(多発性)、扁平上皮癌に関与。
この遺伝子はマウスp(pink-eyed dilution)遺伝子のヒトホモログをコードする。コードされているタンパク質は、低分子の輸送に関与する膜タンパク質で、特にメラニン合成の前駆体であるチロシンに関与すると考えられている。この遺伝子は哺乳類の色素形成に関与しており、皮膚の色の変化を制御し、褐色または青色の目の色の決定因子として働くと考えられている。この遺伝子に変異があると2型眼皮膚アルビニズムとなる。選択的スプライシングにより複数の転写産物の変異が生じる。2014年7月、RefSeqより提供。
OCA2遺伝子の発現
皮膚(RPKM 1.8)、甲状腺(RPKM 1.2)、その他8組織で発現に偏りあり
OCA2遺伝子と関係のある疾患
※OMIIMの中括弧”{ }”は、多因子疾患または感染症に対する感受性に寄与する変異を示す。[ ]は「非疾患」を示し、主に検査値の異常をもたらす遺伝的変異を示す。クエスチョンマーク”? “は、表現型と遺伝子の関係が仮のものであることを示す。エントリ番号の前の数字記号(#)は、記述的なエントリであること、通常は表現型であり、固有の遺伝子座を表さないことを示す。
[Skin/hair/eye pigmentation 1, blond/brown hair] 皮膚髪眼色素沈着1感受性 金髪/茶髪
AR 3
[Skin/hair/eye pigmentation 1, blue/nonblue eyes]皮膚髪眼色素沈着1感受性 青/非青色眼
AR 3
皮膚/毛髪/眼の色素沈着における変異の遺伝的不均一性 複数の遺伝子がヒトの正常な皮膚、毛髪、および/または眼の色素沈着に影響を及ぼす。OCA2遺伝子の変異によって影響を受ける色素沈着の表現型はSHEP1と呼ばれている。SHEP2関連(266300)はMC1R遺伝子座(155555)の変異によって決定され、主に赤毛と色白の皮膚を特徴とする表現型を記述する。SHEP3(601800)はTYR遺伝子(606933)の影響を受けた色素変異を含み、SHEP4(113750)はSLC24A5遺伝子(609802)の影響を受けたものである。SLC45A2遺伝子(606202)およびSLC24A4遺伝子(609840)の変異は、それぞれSHEP5(227240)およびSHEP6(210750)という表現型の関連性をもたらす。KITLG(184745)の発現に影響すると考えられる塩基配列の変異は、SHEP7(611664)の表現型関連となる。SHEP8(611724)はIRF4遺伝子(601900)の変異と関連している。ASIP遺伝子の3-プライム非翻訳領域の多型(600201)はSHEP9との関連(611742)に影響する。SHEP10との関連(612267)はTPCN2遺伝子(612163)の変異からなり、SHEP11(612271)はTYRP1遺伝子(115501)近傍の多型と関連している。
Albinism, brown oculocutaneous/Albinism, oculocutaneous, type II 眼皮膚白皮症(先天性白皮症)2型
AR 3
概要
眼皮膚アルビニズム(OCA)は、常染色体劣性遺伝パターンで遺伝するメラニン生合成のまれな遺伝性疾患群である。異なる遺伝子の変異によって引き起こされる8つのタイプのOCAが認められている。すべてのタイプで皮膚、毛髪、眼の色素沈着が減少または消失するが、臨床的な表現型は疾患の重症度の幅広いスペクトラムに沿って変化する。リソソーム生合成に関与する遺伝子の変異によって引き起こされるOCAには、出血性疾患(Hermansky-Pudlak症候群)や化膿性感染症(Chediak-Higashi症候群)のような全身性の異常を伴うまれなタイプもある。
視力低下、光線過敏症、眼振、斜視などの眼症状は、OCAの最も衰弱させる側面である。これらは、虹彩および網膜のメラニン減少と、発育過程における眼球から脳への視神経線維の誤配向と関連している。皮膚色素の減少は、光線過敏症、皮膚がんリスクの増加、他人と違って見えることによる社会的影響につながる。アルビニズム患者に対する差別は、世界の一部で顕著である。
チロシナーゼ陽性眼皮膚アルビニズム(OCA、II型;OCA2)は、常染色体劣性遺伝の疾患で、皮膚、毛髪、眼においてメラニン色素の生合成が低下する。罹患児は出生時にはOCA I型、すなわちメラニン色素が完全に欠如しているように見えるが、OCA II型の患者のほとんどは年齢とともに少量の色素を獲得する。OCA II型の患者は、視力低下や眼振など、アルビニズムに伴う特徴的な視覚異常を有するが、通常はOCA I型よりも軽度である(Leeら、1994;Kingら、2001)。
OCAⅡ型の表現型は非常に多様である。罹患者の毛髪は加齢とともに黒くなり、色素性母斑やそばかすが見られることがある。アフリカ系およびアフリカ系アメリカ人は、毛髪が黄色く、虹彩が青灰色またはヘーゼル色であることがある。表現型変異の一つである「褐色OCA」は、アフリカ系およびアフリカ系アメリカ人の集団で報告されており、明るい褐色の髪と肌色、灰色から褐色の虹彩を特徴とする。毛髪と虹彩は時間の経過とともに濃くなり、皮膚は日光に当たると日焼けする。アルビニズムの眼球の特徴はすべての変異型に存在する(King et al., 2001)。さらに、明らかな皮膚病変をほとんど認めない、いわゆる「常染色体劣性眼アルビニズム」(例えば、Witkopら、1978およびO’Donnellら、1978を参照)の以前の報告は、現在ではOCA1またはOCA2の表現型スペクトラムの一部である可能性が高いと考えられている(Leeら、1994;Kingら、2001)。
疫学
西洋世界におけるアルビニズムの全体的な有病率は17,000~20,000人に1人と推定され、民族や地域によってかなりの差がある。70人に1人がOCA変異対立遺伝子を持っていると推定されている。アフリカでは、有病率は5000~15000人に1人と推定されているが、1/1400という高さの地域もある。稀な常染色体劣性遺伝性疾患では、両親の近親婚がより頻繁にみられる。この現象はアルビニズムのある集団でよく報告されており、文化的規範や地理的に交流がなく狭い範囲で婚姻を繰り返していることなどが関係している。
眼皮膚アルビニズム2型(OCA2)は、サハラ以南のアフリカにおける高い有病率のため、世界的に最も一般的なアルビニズムの型であり、その有病率はナイジェリアの15,000人に1人という低いものから、ジンバブエの特定の集団の1,000人に1人という高いものまである。アフリカ人では、OCA2はほとんどの場合、創始者効果に関連するOCA2遺伝子の一般的な2.7kbの間質性欠失に起因する。米国におけるOCA2の推定有病率は36,000人に1人である。
分類と用語
「部分」または「完全」アルビニズム、「完全」または「不完全」アルビニズム、「チロシナーゼ陽性」または「チロシナーゼ陰性」アルビニズム、「黄色変異」または「赤褐色」アルビニズムなどの古い用語よりも、原因遺伝子によるアルビニズムの分類が好ましい。眼球皮膚アルビニズム1型(OCA1)から眼球皮膚アルビニズム8型(OCA8)までの8つの非症候群性アルビニズムが認められている。症候性OCAには、11型のHermansky-Pudlak症候群とChediak-Higashi症候群が含まれる。
病因
メラニン生合成の欠損-OCAは、メラニン生成酵素(すなわち、チロシナーゼTYR、チロシナーゼ関連タンパク質1、TYRP1およびメラノソームに存在する特異的輸送タンパク質を含む、メラニン生合成経路に関与するタンパク質をコードする遺伝子の突然変異によって引き起こされる。
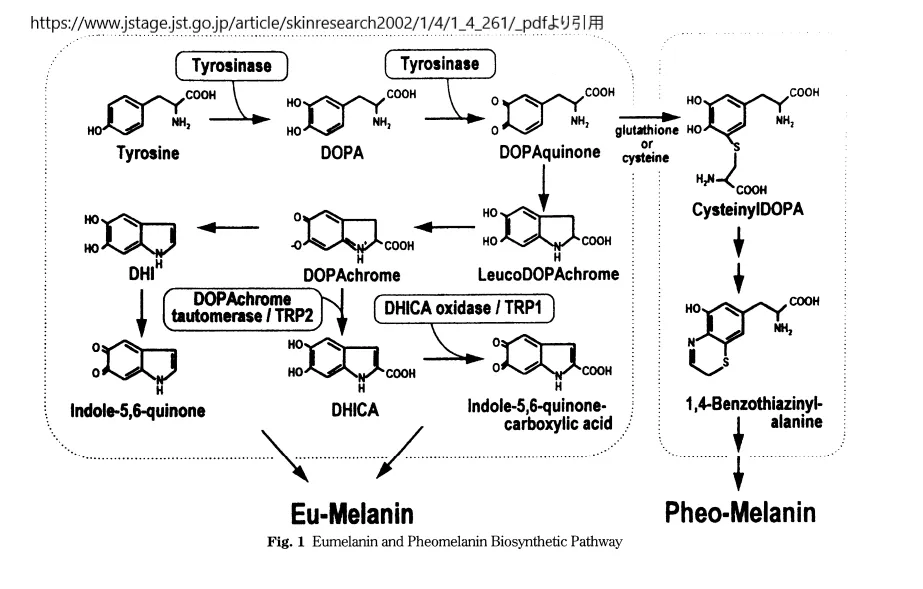
メラニン代謝経路https://www.jstage.jst.go.jp/article/skinresearch2002/1/4/1_4_261/_pdfより引用
毛髪、皮膚、眼の色素沈着は、紫外線を吸収し、日光による皮膚損傷、非黒色腫皮膚がん、黒色腫から身を守る複合色素であるメラニンの酵素的生合成に依存している。TYR酵素は、L-チロシンをDOPA(ジヒドロキシ-L-フェニルアラニン)に酸化することにより、メラニン生合成の第一段階を触媒する。メラニンには、褐色または黒色のユーメラニンと黄色のフェオメラニンの2種類がある。アルビニズムの患者は、一般的にユーメラニンが減少しているが、毛髪中のフェオメラニンは正常レベルである可能性がある。
メラニンの生合成は、メラノサイト内のリソソーム様小器官であるメラノソーム内で区画化される。皮膚の基底層から、成熟した色素を含んだメラノソームが、よく理解されていない細胞間移行プロセスによって、メラノサイトからケラチノサイトに移行し、皮膚に色を与える。メラノソームはIからIVまでの4段階を経て成熟する。I期とII期のプレメラノソームはまだメラニンを含まないが、III期とIV期はメラニン化する。メラニン合成経路のTYRのような酵素を破壊する遺伝子変異は、皮膚、毛髪、眼の色素沈着を減少させる。皮膚外および眼球外の特徴を有するアルビニズムの症候群型は、多くの場合、メラノソームや、Hermansky-Pudlak症候群の血小板密集体やChediak-Higashi症候群のリソソームなど、他の組織におけるメラノソームに関連した重要な小器官の生合成を阻害することにより、色素移動のプロセスを破壊する遺伝子変異に起因する。
ヒトの場合、両眼視には視交叉で両目から反対側の脳に渡る神経線維が必要である。胚発生時に存在するメラニンを含むいくつかの因子が、網膜と血管構造の発達、および網膜の神経節細胞が外側被殻核へ、そして後頭皮質へと向かう経路を誘導している。胚発生時に眼球メラニンが欠乏すると、神経線維の誤った経路と小窩の異常な発達が起こる。視力の低下は、例えば、小窩の形態と小窩の錐体特殊化、眼振、視神経線維のミスルートなどといった、おそらく複合的な要因によるものと考えられる。アルビニズムでは、後虹彩上皮および網膜色素上皮に存在する神経外胚葉由来のメラノサイトが影響を受ける。虹彩の色は、部分的には神経堤由来の間質メラノサイトによって決定される。ルテインやキサントフィルなどの他の色素は網膜に存在し、アルビニズムでは影響を受けない。
眼皮膚アルビニズム2型(OCA2; MIM #203200)はOCA2(以前はPと呼ばれていた)遺伝子の変異によって引き起こされ、この遺伝子はメラニン生成をサポートするpH調節タンパク質をコードしている。
臨床症状
OCAでは表皮および毛包のメラノサイトの数は正常である。眼皮膚アルビニズム1型(OCA1)ではメラノソームの発達が変化している。眼球皮膚アルビニズム1A型(OCA1A)と推定される20週齢の胎児の死後眼球検査では、成熟したメラノソームは通常妊娠7週齢までに存在するのに対し、I期またはII期のメラノソームしか認められず、網膜色素上皮にメラニンが存在しないことが示された。OCA1Aと推定される少年の死後病理所見では、窩洞の欠如と無桿体域が認められた。網膜色素上皮のステージIIIのメラノソームは正常であったが、数は減少していた。OCA1Aと推定される高齢女性の病理組織検査では、小窩の発達が認められず、網膜色素上皮にメラノソームは確認されなかった。OCA1を有する20週齢の胎児の皮膚サンプルの電子顕微鏡検査では、メラノソームはII期までしか認められず、メラニン合成の欠如と一致している。
毛髪と皮膚
OCAの人は、他の家族に比べて、皮膚と毛髪の色素沈着が顕著である。色素沈着は、色素沈着が強い家系では、より容易に認められる。
出生時に白髪やまつ毛のある子供は、眼皮膚アルビニズム1型(OCA1)である可能性が高い。出生時に認められる白髪は、OCA1Aでは白髪のままであるが、OCA1Bでは時間の経過とともに濃くなることがある。OCA1Aの人は、シャンプーや水のミネラルによって毛髪がわずかに黒くなるように見えるが、眉毛やまつ毛は白いままである。毛髪の灰色または銀色の光沢はチェディアック東症候群に特徴的である。
OCA2の患者は通常、金髪または赤みがかった金髪で生まれる。OCA3はアフリカ系ではルファス(赤色)または褐色のOCA表現型を示す。彼らは、銅赤色(赤褐色)の皮膚、ジンジャー色(黄赤色)の毛髪、希薄な虹彩色、または明るい褐色の毛髪と皮膚、青緑色から褐色の眼を持つ。白人のOCA3患者は、臨床的にはOCA2患者と類似している。
加齢に伴い、日光に曝された皮膚は荒れ、厚くなり、光線性角化症が増加する。ある程度のメラニン合成が可能な患者では、日光黒子(そばかす)が発現する。
皮膚癌のリスク
OCA患者では、10代までに発症する可能性のある早期皮膚癌のリスクが高い。扁平上皮癌はOCA患者に発生する最も一般的なタイプの癌ですが、基底細胞癌もある。黒色腫はまれであり、生命を脅かす可能性があると考えられているものの、色素がないため診断は困難であり、高い疑い指数を必要とする。色素沈着の低下により、黒色腫はしばしばピンク色または赤色(無色素性)病変として現れるため、診断時に認識することが難しく、進行していることが多い。OCAにおける黒色腫のほとんどは、背部または下肢に発生すると報告されている。
診断
OCAに対する認識を高めることは、早期診断と早期管理のために必要であり、視力を最大限に発達させる最良の機会を提供する。
臨床診断
OCA の臨床診断は、身体診察および眼科的総合検査における以下の所見の有無に基づく。
眉毛やまつ毛を含む皮膚や毛髪の色素沈着。虹彩の色素沈着が減少し、眼の色はピンクから青、緑、灰色、または淡褐色。
眼科的検査で検出される特徴的な眼の変化と視力異常。これには、羞明、眼振、虹彩経光、窩洞低形成、視力低下などがあり、すべてのタイプの OCA に共通する。
色素性アルビニズムの患者(眼球皮膚アルビニズム1A型を除くすべてのタイプ)は、臨床表現型に著しい重複を示し、アルビニズムのタイプの臨床診断を困難にしている。これらの患者では、分子診断が可能であれば、正確な診断のためだけでなく、正確な遺伝カウンセリングや生殖リスクの評価のためにも重要である。
例えば、出血性疾患、肺症状、再発性感染症といった関連する全身症状の存在は、Hermansky-Pudlak症候群やChediak-Higashi症候群などの症候性OCA亜型の診断を示唆する。
眼科的検査:OCA が疑われる患者では、総合的な眼科的検査により、診断の根拠となる一連の所見が発見される。
すべてのタイプの OCA で認められる変化には以下が含まれる。
・照度を下げ、最良の屈折矯正を行っているにもかかわらず、視線が合わない、年齢相応の視力が低下していると感じることが多い。
・眼振(水平性振戦/突進性眼振、回転性/捻転性眼振、周期性交互眼振)は、代償的な頭部姿勢の有無に関わらず、典型的に認められる。頭部姿勢は眼振を減衰させ(ヌルゾーンと呼ばれる)、視力を改善する。時に、ヌルゾーンは主注視下にあり、好ましい頭部姿勢は特定されない。眼振は輻輳によっても減弱する(例えば、近距離を見る場合に減弱するなど)。アルビニズムの同胞は、一般的に眼の構造は類似しているが、視力は不一致であることがある。
・高い屈折異常(遠視、近視、乱視)がよくみられる。
・外斜視(発散性アライメント)、内斜視(交差性アライメント)、および/または垂直性アライメントを含む斜視(眼球のズレ)は、交互カバー検査でよく検出される。アルビニズムでは、視軸が瞳孔中心に対して鼻側に位置することが一般的であるため、等方視はプリズム検査や代替カバー検査で測定されるよりも小さく見え、外斜視はプリズム検査で測定されるよりも大きく見える。
・OCA患者では、斜視に起因する立体視(細かい奥行き知覚)の欠如や低下がよくみられ、臨床での標準的な検査で測定することができる。しかし、アルビニズムの場合、視交叉での網膜視交叉神経線維の過剰な交差のため、眼がまっすぐであっても立体視は低下または欠如する。通常、立体視は欠如しているが、視力の良い患者でも立体視が低下している場合がある。
・細隙灯検査では、瞳孔の開いていない虹彩全体を観察するのに十分な倍率の小さな明るい光を用いると、通常、ある程度の虹彩の透過光が認められる。虹彩の色は、アルビニズムがあるかどうかの敏感な指標ではない。「ピンク色」の虹彩は、特定の照明条件下でスリットランプなしで検出できる、ほぼ完全な虹彩の透過照明を指す。虹彩の透過照明はアルビニズムに特有のものではない。網膜の色が薄い(すなわち、赤色より黄色またはオレンジ色)ことも認められる。
・拡大眼底検査では、少なくともある程度の窩洞低形成が認められる。視力の良好な一部の患者では黄斑に環状反射がみられ るが、umboは通常同定されない。光干渉断層計(OCT)は、この所見を支持する黄斑の迅速な体積画像を提供する。スペクトル領域OCT技術を用いた黄斑低形成の等級付けは非侵襲的であり、視力予後の推定が可能である。しかし、窩洞形態がほぼ正常でも視力が低下するものがあり、アルビニズムの視力障害には他の要因が関与していることが示唆される。脈絡膜血管は黄斑で可視化される場合とされない場合があり、正常な小窩の血管非透過帯は一般的に欠如している。網膜色素上皮のメラニン色素は一般に欠如しているが、黄斑に粒状メラニン色素が認められることがあり、比較的良好な視力と関連している。視神経は通常、軽度の視神経低形成のため、乳頭周囲にわずかなハローを有し、陥凹は最小限か全くない。
・視覚誘発電位(VEP)を用いると、過剰な網膜散 列による網膜線維の誤走行を確認することができる。分子検査が利用しやすくなったため、アルビニズムの診断を確定するためにVEPを使用する頻度は減少している。過剰な除脈は、機能的磁気共鳴画像法および陽電子放射断層撮影法でも示されている。
分子診断
異なるタイプのOCAは表現型がかなり重複しているため、正確な診断には多遺伝子パネルまたは包括的ゲノム配列決定を用いた分子検査が一般的に好ましく、ほとんどのタイプのOCAで利用可能である。
すべての常染色体劣性遺伝性疾患と同様に、関与する遺伝子の2つの変異コピー、それぞれ反対側の対立遺伝子が同定されなければならない。しかし、複合ヘテロ接合体である罹患者では、現在の技術では1つの変異しか検出できない可能性があるため、遺伝子検査の結果は示唆的ではあるが結論には至らないことがある。検出できない変異は、関連するOCA遺伝子のDNA配列決定や、これらの遺伝子の重複や欠失の評価ではカバーされない領域にある可能性が高い。このような場合、診断は臨床的特徴のみに基づいて行われる。OCA の臨床的特徴を有するが、遺伝子検査が陰性である患者の中には、未確認の他のタイプの OCA が存在する可能性がある。OCAの二遺伝性遺伝(digenic inheritance)を示す証拠はない。
分子生物学的検査は、患者管理、サーベイランス、予後、遺伝カウンセリング(例えば、 家族計画)に影響を与える可能性がある。Hermansky-Pudlak症候群の正確な分子診断は、特定の型に関連する臨床症状に基づいて、医学的管理およびフォローアップの推奨を変更することにより、転帰に影響を与える可能性がある。
修飾遺伝子
Chiangら(2008)は、OCA2遺伝子の複合ヘテロ接合体変異によるOCA2を有するヒスパニック系の女児を報告した。彼女は青白い皮膚、青い虹彩、水平眼振、光を透過する虹彩、小窩反射の欠如、アルビン眼底、視力低下などの視覚障害を有していた。しかし、OCA2には珍しい赤金髪の巻き毛があった。罹患していない母親はプエルトリコ系とキューバ系、罹患していない父親はドミニカ系とエクアドル系であった。それぞれの親はOCA2突然変異のヘテロ接合体であった。さらに遺伝子解析を行ったところ、女児とその父親にTYRP1遺伝子のヘテロ接合体変異(S166X;115501.0002)が同定された。父親は、OCA2遺伝子座とTYRP1遺伝子座のハプロイン不全を併せ持つが、顕著な表現型を示さなかった。赤毛(SHEP2;266300)に関連するMC1R遺伝子(155555)の変異は同定されなかった。Chiangら(2008)は、TYRP1のハプロ不全はOCA2の表現型を修正し、MC1Rの赤対立遺伝子がない場合に赤毛をもたらすと結論づけた。
集団遺伝学
Leeら(1994)は、米国におけるOCA2の全頻度を約1:36,000としたが、アフリカ系アメリカ人では約1:10,000であり、ナイジェリアのイボ族では1:1,100の有病率(Okoro, 1975)、南アフリカのネグロイドでは約1:3,900の有病率(Kromberg and Jenkins (1982, 1984))と言われており、このグループの最も一般的な劣性遺伝疾患である。サハラ以南のアフリカでは、OCA2は多くの罹患率の原因となっており、皮膚癌と視覚障害が重要な後遺症である。
KagoreとLund(1995)は、ジンバブエの首都ハラレの学童におけるOCA2の有病率は1:2,833であることを明らかにした。この有病率に基づき、OCA2の遺伝子頻度は0.0188、保因者頻度は1:27と推定された。アルビニズムの学童のほとんどは、多数派のショニー民族に属していた。ショニー族の文化では近親婚が推奨されていないため、この高率はおそらく、移動が限られた比較的小さな集団における遺伝的ドリフトの結果であろう。
Stevensら(1997)はOCA2染色体780本中10本(1.3%)に2.7kbのP遺伝子欠失(611409.0001)を発見した。OCA2陽性者では、アフリカ南部のOCA2染色体170本中131本(77%)、ザンビアのOCA2染色体14本中11本(79%)、中央アフリカ共和国のOCA2染色体12本中4本(33%)に欠失が認められた。この研究により、欠失対立遺伝子がアフリカ起源であることが確認された。ハプロタイプ解析から、欠失変異は一度しか起こらず、これらのアフリカの集団が分岐する前(約2,000〜3,000年前と推定される)に生じたことが示唆された。OCA2変異、特に2.7kbの欠失の頻度が異常に高いことから、選択あるいは遺伝的ドリフトが示唆された。
アルビニズムの高い頻度は、いくつかのアメリカ先住民の間で見つかっており、その頻度はJemez族の1:140からNavajo族の1:3,750まで様々である(Woolf, 1965; Woolf and Dukepoo, 1969)。Yiら(2003)は、主にアリゾナ州北東部に住むナバホ族のアルビニズムを研究した。ナバホ族におけるアルビニズムの表現型は、OCA2およびOCA4 (606574) の表現型と重なり、それぞれPおよびMATP (SLC45A2; 606202) 遺伝子の突然変異によって引き起こされる。その結果、Yiら(2003)はこれら2つの遺伝子の変異スクリーニングを行った。MATP遺伝子には変異は認められなかったが、アルビニズムを持つナバホ族はすべて、P遺伝子のエクソン10から20までを含む122.5kbのゲノムDNAをホモ接合性で欠失することが判明した(611409.0008)。この欠失対立遺伝子は、他のアメリカ先住民を起源とする34人のアルビニズム患者には見られず、他の民族グループでも報告されていなかった。欠失対立遺伝子の分子的特徴から、Yiら(2003)は3-primer PCRシステムをデザインし、ナバホ集団における保因者頻度を推定した。ナバホ族におけるOCA2の推定有病率は1:1,500から1:2,000である。彼らは、この突然変異は400〜1,000年前の単一の創始者に由来すると推定した。
Suzukiら(2003)は、40人のOCA1陰性の日本人アルビノ患者の突然変異解析を行い、6人の無関係な患者において6つの異なる新規突然変異を同定した。彼らは日本人アルビノ集団におけるOCA2の頻度を8%と推定し、OCA2はOCA1ほど一般的ではないことを示した。34人の患者が未分類のOCAとして残り、第3の遺伝子座がOCAの主要な原因であるかもしれないという考えを支持した。
Rooryckら(2011)は、OCA2遺伝子の184kbの欠失(611409.0015)を、ポーランドの血統を持つ非血縁のOCA2患者3人における創始者変異として同定した。配列解析の結果、2つの切断点は多数のAluおよびL1リピートを含むリピートに富む領域に位置しており、分子機構として非相同末端接合(NHEJ)が示唆された。
この記事の著者:仲田洋美(医師)

この記事の筆者:仲田洋美(医師)






