目次

MMADHC遺伝子は、ビタミンB12(コバラミン)を細胞内の正確な目的地へ届ける「輸送シャペロン」として働く、コバラミン代謝の要となる遺伝子です。この遺伝子の両アレルに変異が生じると、変異の位置に応じてメチルマロン酸血症・ホモシスチン尿症・またはその両方を引き起こす「cblD型細胞内コバラミン代謝異常症」が発症します。同じ遺伝子への変異なのに変異部位によって病態がまったく異なるという独特の遺伝子型-表現型相関を持ち、2025年のPNAS論文ではその分子メカニズムが原子レベルで解明されました。
Q. MMADHC遺伝子とはどのような遺伝子ですか?まず結論だけ知りたいです
A. 第2染色体2q23.2に位置し、細胞内でビタミンB12(コバラミン)をミトコンドリアと細胞質の両方へ正確に振り分ける「輸送シャペロン」として機能する遺伝子です。両アレルに変異が生じると、変異の位置に応じてメチルマロン酸血症のみ(cblD-MMA)・ホモシスチン尿症のみ(cblD-HC)・両方(cblD-MMA/HC)という3つの病態のいずれかを引き起こします。
- ➤遺伝子の基本情報 → 第2染色体2q23.2・8エクソン・296アミノ酸・全身25以上の組織で普遍的に発現
- ➤タンパク質の特徴 → 酵素活性を持たない「修飾ニトロレダクターゼフォールド」を持つ世界初の輸送専用シャペロン
- ➤3つのサブタイプ → 変異部位で病態が完全に異なるという極めて稀な遺伝子型-表現型相関
- ➤臨床症状 → 新生児期の致死的代謝クリーゼから成人期の神経精神症状まで幅広い発症スペクトル
- ➤診断 → 血漿MMA・総ホモシステイン・アミノ酸分析+MMADHC遺伝子解析で確定
- ➤治療の最前線 → ヒドロキソコバラミン・ベタイン療法に加え、AAV遺伝子治療・mRNA-LNP療法が臨床段階へ
1. MMADHC遺伝子の基本情報と疾患の概要
MMADHC(Metabolism of Cobalamin Associated D)は、ヒト第2染色体長腕2q23.2領域に位置する遺伝子です。GRCh38アセンブリ上では149,569,637〜149,587,775 bpの領域にマッピングされ、8エクソンで構成されています。公式シンボルのほかC2orf25・cblD・HMAD・MACD・MAHCDとも呼ばれます。系統学的にはMMADHCのオーソログが真核生物から哺乳類まで広く保存されており、生命維持における基本的な役割の重要性を示しています。
💡 用語解説:コバラミン(ビタミンB12)とは
コバラミン(ビタミンB12)はヒトが自ら合成できない唯一の微量元素補酵素で、細胞内で2種類の活性型補酵素に変換されます。ミトコンドリア内でMUTの補酵素となる5′-デオキシアデノシルコバラミン(AdoCbl)と、細胞質内でメチオニン合成酵素の補酵素となるメチルコバラミン(MeCbl)です。どちらが欠乏するかによって蓄積する有害物質が異なります。MMADHCはこの「振り分け作業」そのものを担う遺伝子です。
組織発現プロファイルを見ると、MMADHC遺伝子は全身25以上の主要組織で普遍的に発現しており、特に骨髄(RPKM 49.1)・食道(RPKM 43.5)で高い発現レベルが確認されています。また胎生期においても、腸管・頭部間葉・中腎・脊索などの初期発生構造での発現が確認されており、正常な個体発生にも不可欠な役割を担っています。進化の過程で生じた機能を持たない偽遺伝子(MMADHCP1)が第11染色体およびX染色体上にも存在します。
💡 用語解説:cblD型とは——「cbl」のアルファベット分類
「cbl」は「cobalamin(コバラミン)」の略で、細胞内コバラミン代謝経路の遺伝的欠陥にA〜Jなどのアルファベットが割り当てられています。cblD型はMMADHC遺伝子変異による病型で、正式名称は「メチルマロン酸血症およびホモシスチン尿症cblD型」。関連する型としてcblC型・cblF型・cblJ型・cblX型などが存在し、それぞれ異なる遺伝子が原因です。
細胞内コバラミン代謝経路には少なくとも7つの遺伝子座が関与しており、MMADHCタンパク質はミトコンドリア経路と細胞質経路への分岐点という最も重要な位置に存在しています。リソソームからのコバラミン放出に関わるABCD4遺伝子(ATP結合カセットサブファミリーDの一員)によるcblJ型とは、代謝経路上の位置づけが大きく異なります。
2. MMADHCタンパク質の三次元構造と「輸送専用シャペロン」への転用
MMADHC遺伝子がコードするのは296アミノ酸からなるタンパク質で、細胞質とミトコンドリアの両方に局在する「デュアルローカリゼーションタンパク質」です。この二重局在性こそが、コバラミンを必要に応じて2つの目的地に届けることを可能にする機能的な基盤です。
精密に区画化された機能ドメイン
🔵 N末端側(1〜115アミノ酸)
ミトコンドリア輸送に必須の領域。Met62〜Met116の配列がミトコンドリアへの標的化シグナルを担い、この部位が破壊されるとAdoCblの合成が不可能になります。細胞質経路(MeCbl合成)には不要な領域です。
🔴 C末端側(116〜296アミノ酸)
両経路の基盤構造を形成。特にアミノ酸残基197〜226・246〜259の領域は細胞質経路(MeCbl合成)の制御に強い影響を持ちます。MMACHCタンパク質との結合インターフェースもここに位置します。
💡 用語解説:シャペロン(Chaperone)とは
シャペロンとは、他のタンパク質や分子が正しく折り畳まれたり、正確な場所に届けられたりするのを手助けする「付き添い役」のタンパク質です。MMADHCの場合は、反応性が高く不安定なコバラミン誘導体を安全に保護しながら、ミトコンドリアまたは細胞質の標的酵素のそばまで確実に運ぶ「細胞内宅配便」の役割を果たします。
構造生物学が解き明かした「転用(リパーパシング)」の奇跡
X線結晶構造解析と小角X線散乱(SAXS)によって、MMADHCのC末端領域が2.2Åの高解像度で構造決定されました。最も驚くべき発見は、MMADHCのC末端部分が「修飾ニトロレダクターゼフォールド(modified nitroreductase fold)」と呼ばれる立体構造を採用していながら、既知の酵素活性が一切ないという事実です。
💡 用語解説:ニトロレダクターゼフォールドと「転用(リパーパシング)」
「ニトロレダクターゼフォールド」とは本来、酸化還元酵素に特有の触媒ドメインの立体構造です。MMADHCはこの構造を持ちながら酵素活性は完全に失われており、MMACHCと結合するための足場として「転用(リパーパシング)」された、世界で初めて同定された事例です。MMACHCとは配列の保存性が低いにもかかわらず、驚くべき構造的相同性が確認されています。

3. MMACHC–MMADHC複合体の物理化学的メカニズム
細胞質におけるコバラミンの処理と輸送は、MMACHC・MMADHC・メチオニン合成酵素還元酵素(MTRR)・メチオニン合成酵素(MTR)から構成される多タンパク質複合体内で進行します。このカスケードの中でMMACHCとMMADHCの相互作用は、コバラミンをミトコンドリアまたは細胞質の標的酵素へと振り分ける最も決定的なステップです。
コバルト–硫黄(Co–S)配位結合という「分子的留め金」
2025年にPNASで発表された最新研究では、MMACHC–MMADHC複合体の3.4Å解像度の結晶構造が解明されました。その核心的な発見は、MMADHCがアミノ酸残基254〜266で構成されるβヘアピン構造をMMACHCの活性部位に挿入し、自身のCys-261(システイン261番残基)の硫黄原子をコバルトイオンへの上部アキシャル配位子として提供するという機構です。
💡 用語解説:Co–S配位結合(コバルト–硫黄配位結合)とは
コバルト(Co)イオンはコバラミンの中心金属です。MMADHCのCys-261にあるシステイン残基の硫黄(S)原子がこのコバルトイオンに強く配位(電子対を提供して結合)することで、2つのタンパク質を強固につなぐ「分子的留め金(molecular clasp)」が形成されます。複合体内でのCo–S結合長は約2.5Åで、不安定なチオラト-コバラミン誘導体が自然分解するのを防ぎ、標的酵素への安全な輸送が完了するまでコバラミンを保護します。
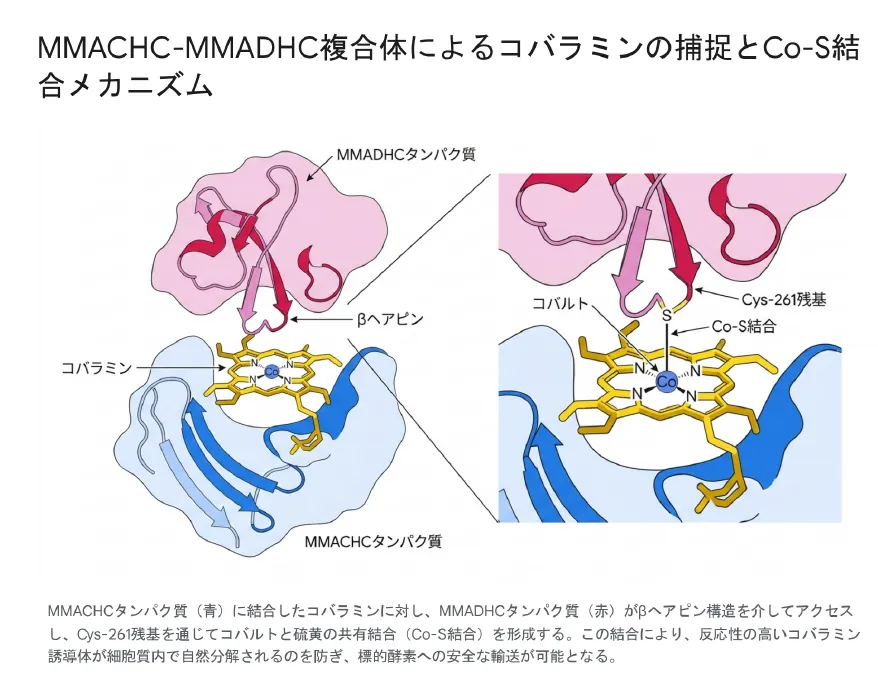
MMACHCタンパク質(青)に結合したコバラミンに対し、MMADHCタンパク質(赤)がβヘアピン構造を介してアクセスし、Cys-261残基を通じてコバルト–硫黄の配位結合(Co–S結合)を形成する。この結合により反応性の高いコバラミン誘導体が細胞質内での自然分解から保護され、標的酵素への安全な輸送が可能となる(2025年PNAS論文より)。
臨床的に同定されているMMACHCの81種類・MMADHCの11種類のミスセンス変異を結晶構造上にマッピングした結果、タンパク質間境界面に位置する変異は一つも存在しないことが明らかになりました。これは高親和性の複合体形成を駆動する一次的な力が、アミノ酸側鎖間の物理的相互作用ではなく、圧倒的にCys-261を介したCo–S配位結合にあることを明確に裏付けています。共鳴ラマン分光法による解析でCo³⁺の酸化状態が確認されており、標的酵素への受け渡しまでコバラミンを安全に保持する精巧な保護機構であることが実証されています。
4. 遺伝子型と3つの生化学的サブタイプ
MMADHC遺伝子変異の最も重要な臨床的特性は、変異が遺伝子上のどの位置に生じるかによって、まったく異なる3つの生化学的病態が引き起こされることです。「同じ遺伝子の変異なのになぜ全く違う病気になるのか?」という疑問への明確な答えがここにあります。
🔵 cblD-MMA(MMA単独型)
変異部位:N末端側(Met116より上流)のナンセンス変異・フレームシフトなどの機能喪失型変異
欠乏補酵素:AdoCblのみ
MMA↑↑・tHcy正常・Met正常。ミトコンドリア経路のみ障害され、細胞質経路は正常機能します。
🟢 cblD-HC(HC単独型)
変異部位:C末端保存領域(p.D246〜L259)のミスセンス変異(βヘアピン近傍)
欠乏補酵素:MeCblのみ
MMA正常・tHcy↑↑・Met低下。細胞質経路のみが特異的に阻害されます。
🔴 cblD-MMA/HC(複合型)
変異部位:C末端広範ナンセンス変異・大規模欠失・20アミノ酸以上の切断
欠乏補酵素:AdoCbl+MeCbl両方
MMA↑↑・tHcy↑↑・Met低下。両経路が遮断される最重症の生化学的プロファイル。
| サブタイプ | 主な変異形式 | MMA | tHcy | Met |
|---|---|---|---|---|
| cblD-MMA | N末端ヌル変異(Met116上流) | ↑↑著明上昇 | 正常 | 正常 |
| cblD-HC | C末端ミスセンス(D246〜L259) | 正常 | ↑↑著明上昇 | ↓低下 |
| cblD-MMA/HC | C末端広範ヌル・20aa以上切断 | ↑↑著明上昇 | ↑↑著明上昇 | ↓低下 |
MMADHC遺伝子変異に基づくcblD型コバラミン代謝異常症の遺伝子型–生化学的表現型相関
5. 発症時期と臨床症状のスペクトル
cblD型代謝異常症の臨床症状は極めて不均一で、生化学的サブタイプ・残存機能・発症年齢によって多様なスペクトルを示します。発症は胎児期から成人期に至るまであらゆるライフステージで起こり得ます。
🍼 胎児期・新生児期
最重症例では子宮内で非免疫性胎児水腫・心筋症・子宮内発育遅延として発症します。出生後早期には小頭症・重度哺乳不良・筋緊張低下・進行性脳症、ケトアシドーシスや高アンモニア血症を伴う致死的な急性代謝クリーゼを呈することがあります。
👶 乳児期・幼児期(1〜3歳)
体重増加不良(Failure to Thrive)・成長障害が顕著になります。神経学的異常として点頭てんかんを含む難治性けいれん発作・全般的発達遅滞が現れます。巨赤芽球性貧血や血球減少症も一般的に認められます。
🧑 青年期・成人期(遅発型)
小児期を大きな問題なく経過した軽症患者でも、記憶障害・幻覚・精神病様エピソード・認知症が発現することがあります。亜急性連合性脊髄変性症による下肢のしびれ・歩行困難も引き起こされます。
🧬 代謝ストレス時(任意年齢)
感染症・絶食・手術などをきっかけに急性代謝クリーゼが誘発されます。MMA蓄積サブタイプでは大脳基底核(淡蒼球)の両側性ラクナ梗塞(代謝性ストローク)を来たし、不可逆的なジストニアを残すことがあります。
蓄積代謝産物によって異なる臓器障害パターン
📌 MMA蓄積による病態
- 大脳基底核の代謝性ストローク・不可逆性ジストニア
- 腎毒性→尿細管性アシドーシス→慢性腎不全
- 拡張型または肥大型心筋症
- 膵炎・肝脂肪変性
- 免疫機能不全
📌 ホモシステイン蓄積による病態
- 生命を脅かす血栓塞栓症(腎動脈血栓症・肺高血圧症)
- 非定型溶血性尿毒症症候群(aHUS)
- 血栓性微小血管障害症(TMA)
- 全身血管内皮毒性による血管障害
6. 診断アルゴリズムと新生児スクリーニングの課題
cblD型の診断は、血液・尿中の生化学的マーカーの複合的評価から始まり、最終的に分子遺伝学的検査によって確定されます。
🔬 尿中有機酸(UOA)・血清MMA
メチルマロン酸を定量。副代謝産物として3-ヒドロキシプロピオン酸・メチルクエン酸・チグリルグリシンが検出されることがあります。
🔬 血漿総ホモシステイン(tHcy)
ホモシステイン蓄積を評価。採血後の血清分離遅延により1時間あたり最大10%人為的に上昇する「アーティファクト」に注意が必要です。
🔬 血漿アミノ酸(PAA)
低メチオニン血症の確認。メチオニンが逆に上昇するシスタチオニンβ合成酵素(CBS)欠損症との鑑別に重要です。
🔬 血中ビタミンB12レベル
後天性の栄養性ビタミンB12欠乏や母体由来の二次的欠乏を除外するために必須の検査です。
生化学的所見がcblD型のいずれかのサブタイプに合致した場合、次世代シーケンサー(NGS)を用いたMMADHC遺伝子の全コード領域解析および欠失/重複分析によって確定診断とします。
💡 用語解説:新生児マススクリーニングの「盲点」と二次スクリーニング
新生児スクリーニングは乾燥濾紙血(DBS)を用いたタンデム質量分析(MS/MS)でプロピオニルカルニチン(C3)上昇を捕捉します。cblD-MMAおよびcblD-MMA/HCはC3上昇で検出できますが、cblD-HCではC3が上昇しないため見逃されます。この盲点を補うべく、DBSでMMAとtHcyを直接定量する「二次スクリーニング」が導入されつつあります。2025年イタリアのデータでは偽陽性率が0.084%まで低下し、陽性的中率は8.4%(後天性B12欠乏を含めると57%)に改善したことが示されています。
7. 治療と長期管理:標準プロトコルと次世代療法
MMADHC変異を根本的に修復する治療薬は現時点では承認されていませんが、国際ガイドライン(Huemer et al., 2017等)に基づく早期かつ積極的な介入により生存率と予後を大きく改善することが可能です。日本ではメチルマロン酸血症は指定難病246に認定されており、医療費の公的補助が提供されています。
急性代謝クリーゼの管理
感染症や絶食を契機とした急性代謝不全では、ブドウ糖を含む静脈内輸液を速やかに開始しタンパク質摂取を一時的に中止、代謝専門医の指導下で非経口的なヒドロキソコバラミン(OHCbl)の投与を直ちに開始します。重篤なアシドーシス・高アンモニア血症・腎不全が進行した場合は持続的血液濾過透析も必要となります。乳幼児では一晩以上の絶食を厳格に回避しなければなりません。
長期維持療法の主要薬剤
| 薬剤名 | 推奨用量 | 投与頻度 | 標的パラメータ | 根拠 |
|---|---|---|---|---|
| ヒドロキソコバラミン(OHCbl) 筋注/皮下/静注 |
0.3 mg/kg/日または1 mg/日 | 毎日(安定後は漸減) | MMA・tHcy低下、血漿B12維持 | 確立 |
| ベタイン(経口) | 250〜300 mg/kg/日 | 1日3回分割 | tHcy低下・Met正常化 | 確立 |
| L-カルニチン(経口) | 50〜100 mg/kg/日 | 1日3回分割 | 毒性有機酸の排泄促進 | 一部確立 |
| ホリナートカルシウム/葉酸(経口) | 5〜15 mg/日 | 1日2〜3回分割 | 葉酸サイクルの補完 | 理論的 |
cblD型代謝異常症の長期維持療法における主要薬剤(Huemer et al. 2017ガイドライン等に基づく)
💡 用語解説:ヒドロキソコバラミン vs シアノコバラミン
市販のビタミンB12サプリメントに含まれる「シアノコバラミン」と、治療に使用する「ヒドロキソコバラミン(OHCbl)」は別物です。OHCblはシアノコバラミンと比較して細胞内での保持率が高く、活性型への変換効率が優れています。cblD型の治療においては診断の確定を待たず、再メチル化障害が疑われる時点でのOHCbl即時投与がガイドラインで強く推奨されています。一般のサプリメントでは代替できません。
食事管理では、cblD-MMAおよびcblD-MMA/HCの患者でプロピオン原性アミノ酸(バリン・イソロイシン・メチオニン・スレオニン)を多く含む天然タンパク質の摂取制限が必要です。一方、cblD-HC(単独ホモシスチン尿症)の患者においてはメチオニン制限食は有害であるため絶対に行ってはなりません。
次世代治療:遺伝子治療とmRNA療法の最前線
🧬 AAVベクターを用いた遺伝子治療
米国NIH/NCATSはMMAを対象とするAAV遺伝子治療(MMA-101)を2025年秋に第1相臨床試験開始予定。欧州Genespire社は免疫遮蔽レンチウイルスベクター(ISLV)の強力な前臨床データをASGCT 2025年次総会で発表しており、臨床試験への移行が期待されています。
💉 mRNA–脂質ナノ粒子(LNP)療法
Moderna社のmRNA-3705は欠損タンパク質をコードするmRNAをLNPに封入して静脈内投与する治療法。第1/2相臨床試験(NCT03810690)が進行中で、ゲノム組み込みリスクがない安全性の高さから開発が加速しています。
8. 遺伝カウンセリングと保因者スクリーニング
cblD型コバラミン代謝異常症は常染色体劣性遺伝の疾患です。両親はそれぞれ1コピーの変異アレルを持つ保因者(キャリア)であることがほとんどで、通常は症状を示しません。
💡 用語解説:常染色体劣性遺伝とは
「常染色体」とは性染色体(X・Y)以外の染色体のこと。「劣性(潜性)」とは2本の染色体の両方に変異がある場合にのみ症状が現れる遺伝形式です。両親が各1コピーの変異を持つ保因者の場合、子どもが発症する確率は理論上25%(4人に1人)、保因者になる確率は50%、変異を持たない確率は25%です。
- ➤再発リスクの説明:兄弟姉妹が罹患者となる確率は各妊娠につき25%。ご両親の保因者状態を確認した後、祖父母世代への検査拡大も選択肢となります。
- ➤出生前診断の選択肢:次の妊娠において絨毛検査または羊水検査による出生前遺伝子診断が可能です。また重篤な場合は着床前遺伝子検査(PGT-M)も選択肢のひとつです。
- ➤保因者スクリーニング:将来の妊娠を考える方が事前に保因者かどうかを調べるキャリアスクリーニングも有効です。ACMGの推奨では、すべてのカップルへの拡張保因者スクリーニングが推奨されています。
- ➤関連遺伝子との比較:コバラミン代謝に関わるHCFC1遺伝子・THAP11遺伝子(cblX型)、PRDX1遺伝子との関連も含め、臨床遺伝専門医による総合的な遺伝カウンセリングを受けることが重要です。
9. よくある誤解と専門医からのメッセージ
誤解①「ビタミンB12サプリを飲めばよい」
cblD型はビタミンB12の「摂取量不足」ではなく、「細胞内輸送機構の欠陥」が原因です。市販のシアノコバラミンサプリでは治療にならず、ヒドロキソコバラミンの注射による治療が必要です。
誤解②「NBS正常だったから安心」
cblD-HCサブタイプはC3が上昇しないため、標準的な新生児スクリーニングで見逃されます。症状があればスクリーニング陰性でも専門医への相談が重要です。
誤解③「MMAがないcblD-HCは安全」
cblD-HC型ではMMAは蓄積しませんが、高ホモシステイン血症による深刻な血栓リスクと血管内皮障害があります。「MMAがない=軽症」は誤りで継続管理が不可欠です。
よくある質問(FAQ)
🏥 cblD型・コバラミン代謝疾患の遺伝子検査・遺伝カウンセリング
MMADHC遺伝子変異をはじめとするコバラミン代謝疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] NCBI Gene. MMADHC metabolism of cobalamin associated D [Homo sapiens]. Gene ID: 27249. [NCBI Gene]
- [2] Coelho D, et al. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat Genet. 2012;44(11):1152-1155. [PubMed]
- [3] Plesa M, et al. Structural Insights into the MMACHC-MMADHC Protein Complex Involved in Vitamin B12 Trafficking. J Biol Chem. 2011. [PMC4705923]
- [4] An unusual Co–S bond links B12 chaperones in an interprotein complex. PNAS. 2025. [PNAS 2025]
- [5] Stucki M, et al. Molecular mechanisms leading to three different phenotypes in the cblD defect of intracellular cobalamin metabolism. Hum Mol Genet. 2012;21(6):1410-1422. [Oxford Academic]
- [6] Disorders of Intracellular Cobalamin Metabolism – GeneReviews. NCBI Bookshelf. [GeneReviews]
- [7] Huemer M, et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis. 2017;40(1):21-48. [PMC5203859]
- [8] Cobalamin D Deficiency Identified Through Newborn Screening. JIMD Rep. 2019. [PMC6323031]
- [9] Milder Form of Cobalamin C Disease May Be Missed by Newborn Screening. MDPI Metabolites. 2025;11(3):77. [MDPI]
- [10] Personalized Genome-Scale Modeling Reveals Metabolic Perturbations in Fibroblasts of Methylmalonic Aciduria Patients. Mol Genet Metab. 2025. [PMC12340171]
- [11] MMA-101 | National Center for Advancing Translational Sciences (NCATS). [NCATS]
- [12] メチルマロン酸血症(指定難病246). 難病情報センター. [難病情報センター]






