承認済シンボル:CTNNB1
遺伝子名:catenin beta 1
参照:
HGNC: 2514
AllianceGenome : HGNC : 2514
NCBI:1499
Ensembl :ENSG00000168036
UCSC : uc010hia.2
遺伝子OMIM番号116806
●遺伝子のlocus type :タンパク質をコードする
●遺伝子のグループ:Armadillo repeat containing
Beta catenins
Wnt enhanceosome complex
●遺伝子座: 3p22.1
●ゲノム座標: (GRCh38): 3:41,199,505-41,240,443
遺伝子の別名
beta-catenin
catenin (cadherin-associated protein), beta 1
catenin (cadherin-associated protein), beta 1, 88kDa
catenin beta-1
CTNB1_HUMAN
CTNNB
遺伝子の概要
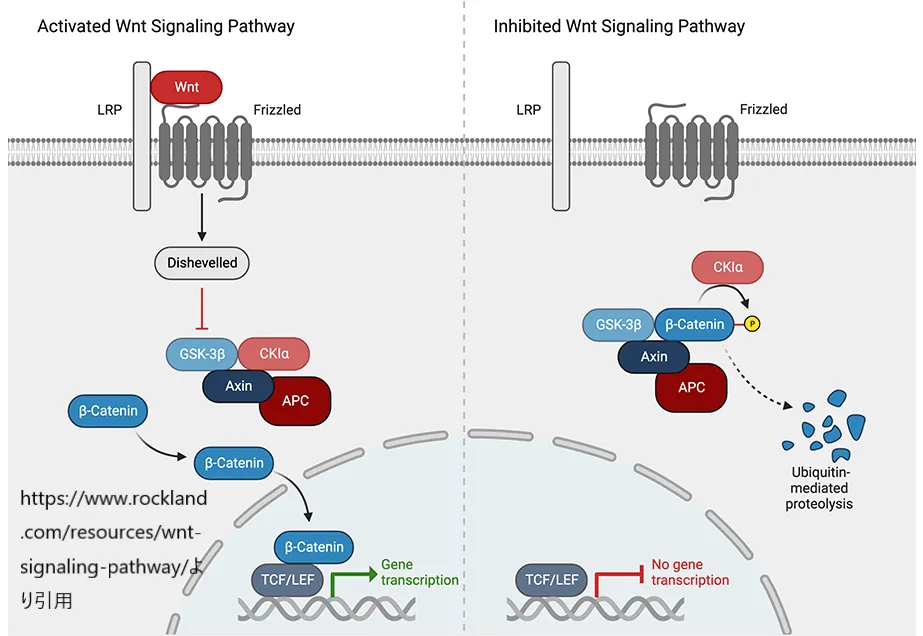
さらに、β-カテニンはWntシグナル伝達経路においても中心的な役割を果たし、細胞の運命を決定する重要な情報伝達メカニズムの一部を形成します。この経路は細胞の成長、分裂、および特殊化を促進し、特に発達初期段階での器官形成や成体の組織再生に必須です。
Wntシグナル伝達が活性化すると、β-カテニンは細胞核内へ移動し、DNA上の特定の遺伝子の転写を促進します。これにより、細胞の挙動に関わる様々な遺伝子の発現が変化し、細胞分裂や分化、生存に関わる重要な過程が調節されます。この経路の調節異常は、がんを含む多くの疾患の発生に関連しています。
加えて、β-カテニンは毛包の発育と機能維持にも関与しており、毛髪の成長周期におけるキープレーヤーの一つです。毛包内のマトリックス細胞でのβ-カテニンの活性は、これらの細胞の増殖と毛髪の成長を促進し、正常な毛髪形成を支えます。β-カテニンのレベルと活性の調節不良は、毛髪の成長障害やその他の皮膚関連疾患を引き起こす可能性があります。
CTNNB1遺伝子の変異やその発現レベルの異常は、細胞の異常な増殖や分化失調を引き起こし、腫瘍形成や発達障害の原因となることが知られています。そのため、β-カテニンおよびWntシグナル伝達経路は、がん研究や組織工学における重要な研究対象となっています。
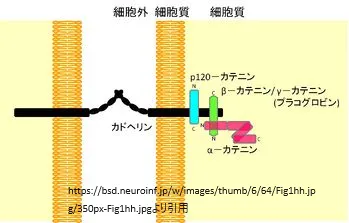
β-カテニンは、細胞間接着の重要な要素であるアドヘレンスジャンクション(AJ)の構成成分です。AJは、上皮細胞が隣接する細胞と結合し、細胞間の物理的な接着を確立することを可能にするタンパク質複合体です。これにより、組織の構造的完全性が保たれ、細胞が適切に配置されます。AJの形成と維持は、臓器の表面を覆う上皮層の構築に不可欠です。
AJは、隣接する細胞間の物理的な接続を仲介することで、細胞同士が一緒に存在するというシグナルを伝達します。また、アクチン細胞骨格に結合することで、細胞形態の維持に寄与します。これらの機能により、AJは細胞の正常な成長、分化、そして行動を調節する役割を果たしています。
細胞の動態、特に胚発生、創傷治癒、腫瘍細胞の転移などの過程では、細胞は上皮を形成し、その後、離れていく必要があります。この過程は、AJの動的な分解と再構築によって精密に制御されています。細胞間の接触が一度確立されると、AJは「接触阻害」と呼ばれる現象を介して細胞分裂を停止させるシグナルを細胞に送ることができます。これにより、細胞の過剰な増殖が防がれ、組織の構造が維持されます。
β-カテニンとAJの研究は、細胞接着、組織の形成、およびがんの転移など、多岐にわたる生物学的過程の理解を深める上で重要な意味を持ちます。これらの知見は、将来的に新しい治療戦略の開発につながる可能性があります。
遺伝子と関係のある疾患
Exudative vitreoretinopathy 7 滲出性硝子体網膜症7 617572 AD 3
Hepatocellular carcinoma, somatic 体細胞性肝細胞がん 114550 3
Medulloblastoma, somatic 体細胞性髄芽腫 155255 3
Neurodevelopmental disorder with spastic diplegia and visual defects 痙性片麻痺と視覚障害を伴う神経発達障害 615075 AD 3
Ovarian cancer, somatic 体細胞性卵巣がん 167000 3
Pilomatricoma, somatic 体細胞性毛母腫(もうぼしゅ) 132600 3
遺伝子の発現とクローニング
細胞質タンパク質には、α-カテニン、β-カテニン、γ-カテニンが含まれ、これらは細胞内シグナル伝達と接着機構において重要な役割を果たします。β-カテニンは、α-カテニンと配列の類似性はないものの、デスモソームに見られるプラコグロビン、およびショウジョウバエの分節極性遺伝子「アルマジロ」の産物と70%のアミノ酸同一性を共有しています。アルマジロは、ショウジョウバエの多タンパク質アドヘレンス・ジャンクション複合体の一部であり、カドヘリンやα-カテニンのホモログを含んでいます。遺伝学的研究から、アルマジロが細胞接着と細胞骨格の整合性に不可欠であることが示されています。
アルマジロ遺伝子は、ショウジョウバエ胚のクチクラパターン形成を制御するセグメント極性遺伝子群の一つとして最初に同定されました。この研究は、細胞接着メカニズムと細胞骨格のダイナミクスの理解において重要な基盤を提供し、細胞間の相互作用と組織の構造維持に対する洞察を深めました。これらの分子の研究は、発生生物学、がん生物学、組織修復など、多岐にわたる生物医学的応用につながる可能性を秘めています。
遺伝子の構造
プロモーター領域がGCに富み、TATAボックスを含むことは、この遺伝子が複雑な転写制御メカニズムを持っていることを示唆しています。マウス上皮細胞において、5-プライムフランキング領域をアルカリホスファターゼのレポーター遺伝子に連結することでプロモーター活性が確認されたことは、この領域が細胞特異的な転写の調節に重要であることを意味しています。
β-カテニンの正確な調節は細胞の振る舞いにとって極めて重要であり、その異常は癌を含む多くの疾患に関連しています。CTNNB1遺伝子の遺伝子構造とプロモーター活性に関するこれらの知見は、β-カテニンの機能を理解し、疾患治療への応用を探るための基礎を提供します。
マッピング
CTNNB1遺伝子は細胞間接着およびWntシグナリング経路における重要な役割を担っており、その異常は癌の発生に関連しています。β-カテニンは、APC(アデノーマポリポシスコリ遺伝子)タンパク質と相互作用し、細胞の増殖と分化を制御します。この相互作用は特に大腸癌の発生において重要な役割を果たします。
Trentらは、β-カテニンのようなAPC結合タンパク質がAPC活性の「下流」調節因子であるため、これらのタンパク質の染色体座位が悪性腫瘍における染色体再配列に関与する可能性があると述べています。このように、CTNNB1遺伝子の位置とその機能は、癌の分子生物学と治療戦略の理解において重要な情報を提供しています。
生化学的特徴
α-カテニンの二量化ドメインの結晶構造研究から、α-カテニンが4-ヘリックス束を介して二量化する構造が明らかにされました。さらに、β-カテニンとの相互作用に関する研究から、α-カテニンとβ-カテニン間の結合機構が詳細に解明されています。
一方、β-カテニンはTCF/LEFファミリーの転写因子とも結合し、Wntシグナル伝達経路における転写活性化の調節に関与します。ICATというタンパク質は、β-カテニンとTCF/LEFファミリー転写因子との相互作用を阻害することで、β-カテニンによる転写活性化を選択的に抑制することができます。これは、β-カテニンの機能を細かく調節することが可能であり、細胞接着を損なうことなく転写活性化を阻害する新たながん治療戦略への道を開くものです。
これらの発見は、β-カテニンが細胞のアドヘレンス接合部およびシグナル伝達経路において果たす複雑な役割を明らかにし、細胞生物学およびがん生物学におけるβ-カテニンの理解を深めるものです。
遺伝子の機能
さらに、β-カテニン/TCF経路は、細胞の運命を決定する重要な役割を果たしており、大腸がん細胞におけるその活性の破壊は、細胞周期の停止と細胞分化の誘導に影響を与えることが示されています。MYC遺伝子の発現は、この転換の中心的な役割を果たし、β-カテニン/TCF4活性の変化によって調節されます。
また、β-カテニン/TCF経路は、細胞外マトリックスの成分であるヘパラン硫酸の生合成にも影響を及ぼします。グルクロン酸エピメラーゼ(GLCE)の発現は、β-カテニン-TCF4複合体によってトランス活性化され、ヘパラン硫酸の構造と機能に重要な変化をもたらす可能性があります。
これらの研究成果は、β-カテニン/TCF経路が多様な生物学的プロセスに深く関与しており、この経路の調節が細胞の成長、分化、および癌の発生において重要であることを示しています。特に、この経路の異常は様々な形態のがんの進行に関与しており、将来的ながん治療の標的として注目されています。
Batlleら(2002年)は、大腸癌や陰窩-絨毛軸においてβ-カテニンとTCFがEphB2/EphB3レセプターとエフリンB1の発現を制御し、細胞集団の選別と腸上皮内の秩序を維持することを明らかにしました。
Kawasakiら(2000年)は、ASEFがAPCと直接相互作用し、ASEF、APC、β-カテニンが同じ複合体に存在する可能性を示唆し、これが細胞骨格や細胞移動に関与していることを示しました。
Neishら(2000年)は、病原性サルモネラに暴露された上皮細胞でのβ-カテニンのリン酸化とユビキチン化の阻害を観察しました。
Eastman and Grosschedl(1999年)は、β-カテニンとLEF1/TCFタンパク質の相互作用とその制御に関する理解を深め、Wntシグナル以外の因子がβ-カテニン活性に影響を与えることも論じました。
Kangら(2002年)は、プレセニリン-1がβ-カテニンのリン酸化に関与する新たな経路を示し、これがβ-カテニンの安定性を増加させることを発見しました。
Muraseら(2002年)は、脱分極によるβ-カテニンの樹状突起からスパインへの移動と、これがシナプス形成に与える影響を明らかにしました。
TetsuとMcCormick(1999)は、β-カテニンがサイクリンD1(CCND1)プロモーターからの転写を活性化することを示しました。彼らは、活性化に必要なTCF/LEF結合部位を含むプロモーター配列を特定しました。p21 RASは、ETSやCREBと結合するプロモーター内の部位を通じてサイクリンD1遺伝子の転写をさらに活性化しました。変異型β-カテニンを発現する細胞は、高レベルのサイクリンD1 mRNAとタンパク質を産生しました。結腸癌細胞でTCFのドミナントネガティブ型を発現させると、サイクリンD1の発現が阻害され、細胞周期のG1フェーズでの停止が観察されました。
Linら(2000)は、乳癌細胞においてCCND1がβ-カテニンの標的の一つであり、高いβ-カテニン活性が患者の予後不良と相関し、乳癌における強力で独立した予後因子であることを見出しました。これは、β-カテニンが乳癌の形成および進行に関与し、治療の標的としての可能性を示唆しています。
Van Akenら(2002年)は、網膜芽細胞腫と正常網膜組織でのカドヘリン-カテニン複合体の研究を通じて、網膜芽細胞腫細胞での複合体の不規則な分布と細胞骨格との弱い結合を明らかにし、この複合体が浸潤促進因子として作用することを示しました。
Widlundら(2002年)は、β-カテニンがメラノーマ細胞増殖の重要な制御因子であり、MITFが重要な下流標的であることを同定しました。正統的Wnt経路の破壊がメラノーマ細胞の成長を阻害することを示しました。
Jamoraら(2003年)は、毛包の形態形成における2つの外部シグナル、WNTタンパク質と骨形成タンパク質阻害剤Nogginによってβ-カテニンが安定化され、Lef1が活性化されることを示しました。このプロセスはE-カドヘリンのダウンレギュレーションと卵胞形成を促進します。E-カドヘリンレベルの強制的な上昇が浸潤と卵胞形成を阻害することを発見しました。
Jarvinenら(2006年)の研究は、β-カテニンの安定化がマウスの口腔上皮と歯上皮で過剰な歯芽の形成を引き起こし、これが新しい歯の形成に関与する新しいシグナル伝達センター、エナメルノットの出現を促すことを示しました。この発見は、歯の発生と形態形成におけるWNT/β-カテニンシグナルの役割を強調しています。
Reyaら(2003年)の研究は、造血幹細胞(HSCs)の自己更新と分化過程におけるWNTシグナルの重要性を示しました。WNTシグナルが造血幹細胞のプールの拡大と、造血幹細胞がin vivoでの正常なホメオスタシスに必要であることを明らかにしました。
YuとMalenka(2003年)は、ラット海馬神経細胞培養においてβ-カテニンの増加が樹状突起の発達を促進することを発見しました。これは、β-カテニンが神経発達における重要な調節因子であることを示しています。
Wikramanayakeら(2003年)は、イソギンチャクNematostella vectensisの胚におけるβ-カテニンの役割を研究し、その安定化と核内移行が軸の同一性や生殖細胞層の形成に重要であることを示しました。これは、β-カテニンの機能が進化的に保存されていることを示唆しています。
Kleberら(2005)は、Bmp2がWnt/β-カテニンの感覚運命誘導活性に拮抗し、相乗的に作用してマウス神経堤幹細胞のマーカー発現と多能性を維持することを発見しました。
Brembeckら(2004)は、BCL9-2がβ-カテニンの接着機能と転写機能の切り替えに関与し、β-カテニンのチロシンリン酸化がBCL9-2の結合を促進し、α-カテニンとの相互作用を阻害することを示しました。
Tianら(2004)は、14-3-3-ゼータがβ-カテニンと相互作用し、β-カテニン依存性の転写を増強し、AKTによるβ-カテニンの活性化を促進することを発見しました。
Guoら(2004)は、Wnt4、Wnt14、Wnt16が発育中のマウス滑膜関節で重複して相補的に発現し、Wnt/CTNNB1シグナル伝達経路が滑膜関節形成の初期段階を誘導するために必要かつ十分であることを結論づけました。
Kaplanら(2004)は、β-カテニンが双極性有糸分裂紡錘体を確立する役割を持つことを発見しました。
Kimら(2005年)は、前立腺ガン細胞における転移抑制遺伝子KAI1のダウンレギュレーションにβ-カテニンとレプチンの抑制作用が関与していることを報告しました。
Essersら(2005)は、インスリンや酸化ストレスシグナルによって制御されるFOXO転写因子とβ-カテニンの進化的に保存された相互作用を報告し、酸化ストレス条件下でのFOXO機能制御におけるβカテニンの役割を実証しました。
Shahら(2006年)は、ビタミンD受容体(VDR)の活性化によって、β-カテニンのシグナル伝達と発がん活性を抑制できることを発見しました。逆に、β-カテニンの高レベルは、1,25-ジヒドロキシビタミンD3の転写活性を増強すると報告されています。β-カテニンの効果は、VDRの活性化因子機能-2ドメインとβ-カテニンのC末端の相互作用によるものです。
Noubissiら(2006年)は、β-カテニンがF-boxタンパク質β-TrCP1のmRNAを安定化させること、およびRNA結合タンパク質CRDBPをβ-カテニン/Tcf転写因子の標的として同定しました。CRDBPの過剰発現は、BTRCP1 mRNAを安定化し、細胞内およびin vivoでBTRCP1レベルを上昇させ、その結果、Skp1-Cullin1-F-box protein (SCF) – BTRCP1 E3ユビキチンリガーゼが活性化され、その基質のターンオーバーが加速されました。CRDBPは大腸がん細胞におけるβ-カテニンシグナル伝達によるBTRCP1とc-Mycの誘導に不可欠です。
Parakhら(2006年)は、β-カテニンの分解につながるN末端90アミノ酸を欠いたβ-カテニンの発現が、卵胞刺激ホルモン(FSH)を介するCYP19A1とCYP11A1のmRNAの誘導を有意に増強することを見出しました。SF1によるCYP19A1の転写活性化には、β-カテニンとの機能的相互作用が必要でした。
Mooreら(2008年)は、Mtgr1、Mtg8、Mtg16がヒトTCF4と相互作用し、β-カテニンはこの相互作用を阻害することを示しました。Xenopus胚での発現実験では、MtgファミリーメンバーがWnt依存性の軸形成を阻害しました。
Liuら(2007年)は、Wnt-β-カテニンシグナル伝達が、発生中の菌状プラコードと味蕾細胞で活性化され、βカテニンの優性安定化突然変異が肥大した菌状乳頭と味蕾の過剰生産を引き起こすことを示しました。
Bahmanyarら(2008年)は、β-カテニンの安定化が異所性NEK2の活性化と同様に、中心体の分裂を誘導することを発見しました。
Malanchiら(2008年)は、初期表皮腫瘍において、癌幹細胞が腫瘍発生特性を持つ唯一の細胞であると同定しました。βカテニンシグナル伝達が癌幹細胞の表現型を維持するのに必須であることを発見しました。
Firesteinら(2008年)は、ヒト結腸がん細胞において2つの機能喪失スクリーニングを行い、その結果を結腸がん検体のコピー数変化と比較しました。彼らが同定した遺伝子の一つであるサイクリン依存性キナーゼ-8(CDK8)は、結腸がんの多くで再発性のコピー数増加を示す13q12.13に位置しています。CDK8の発現を抑制することで、CDK8とβカテニンの高活性を持つ結腸がん細胞の増殖が阻害されることが示されました。CDK8キナーゼ活性は、β-カテニン駆動の形質転換およびβ-カテニンの転写ターゲットの発現に必須であることがわかりました。
Morrisら(2008年)は、転写因子E2F1がβ-カテニン/T細胞因子(TCF)依存性転写の強力な特異的阻害因子であり、E2F1によるアポトーシスに寄与することを発見しました。E2F1の調節異常によってβ-カテニン活性がAPC/GSK3非依存的に抑制され、c-MYCなどのβ-カテニンターゲットの発現が減少することが示されました。この相互作用は、大腸腫瘍がRB1を無傷で保持する理由を説明します。CDK8レベルの上昇は、E2F1によるβ-カテニン/TCF依存性転写の阻害から保護することが示されました。
Chassotら(2008年)とTomizukaら(2008年)は、マウスでのRspo1ノックアウト実験から、Rspo1が女性の性決定においてβカテニンとWnt4シグナルの活性化に必要であることを見出しました。この遺伝子の欠如は女性マウスで少なくとも部分的に性転換を引き起こすが、男性ではそうはならないことが示されました。
家族性大腸腺腫症(FAP)では、APC遺伝子の変異によりβ-カテニンが構成的に安定化し、細胞に永続的な分裂促進シグナルを与えます。Kohlerら(2009年)は、APCのβ-カテニン抑制ドメイン(CID)がβ-カテニンのレベルと転写活性を調節するために必要であることを述べています。CIDの活性の喪失は結腸がん細胞株で観察され、CIDを欠くAPCの短い切断型はこの機能を阻害します。
Huangら(2009年)は、化学的遺伝子スクリーニングによりβ-カテニンを介した転写を選択的に阻害する低分子XAV939を同定しました。XAV939はアクシンの安定化を通じてβ-カテニンの分解を促進し、アクシンと相互作用するタンキラーゼ-1およびタンキラーゼ-2を阻害することによって作用します。
Gattinoniら(2009年)は、Wnt/β-カテニンシグナル伝達の誘導がマウスのCd8陽性T細胞を自己複製能力を持つメモリー幹細胞に発達させることを報告しました。一方で、Driessensら(2010年)は、β-カテニン経路がT細胞の記憶表現型を制御しているという証拠を見いださなかったことを報告し、Gattinoniら(2009年)の結果に対する異なる解釈を提供しました。
Manicassamyら(2010年)は、腸管樹状細胞におけるWnt-β-カテニンシグナルが炎症反応と制御反応のバランスを制御することを発見しました。β-カテニンの発現が抑制されると、炎症性腸疾患モデルマウスでの炎症反応が増強されることが示されました。これらの研究は、β-カテニンが多岐にわたる生物学的プロセスと疾患の発生において重要な役割を果たしていることを示しています。
Yangら(2011年)の研究では、ヒトのがん細胞において、EGFRの活性化がPKM1ではなくPKM2の核への移行を引き起こし、PKM2のK433がc-Srcによってリン酸化されたβ-カテニンのY333に結合することが示されました。この相互作用により、両タンパク質がCCND1プロモーターにリクルートされ、HDAC3がプロモーターから除去され、ヒストンH3がアセチル化されることでサイクリンD1が発現する必要があります。PKM2に依存するβ-カテニンの転写活性化は、EGFRによって促進される腫瘍細胞の増殖と脳腫瘍の発生に関与しています。ヒトの膠芽腫検体では、c-Src活性、β-カテニンY333のリン酸化、PKM2の核内蓄積の間に正の相関が見られ、β-カテニンのリン酸化およびPKM2の核内蓄積レベルは神経膠腫の悪性度および予後と相関していました。
Hoffmeyerら(2012年)は、Wnt/β-カテニンシグナリングとテロメラーゼサブユニットTertの発現との間に分子的関係があることを報告しました。β-カテニンが不足しているマウス胚性幹細胞はテロメアが短く、活性化型β-カテニンを発現する細胞ではテロメアが長くなります。β-カテニンは、多能性転写ネットワークの中心的な要素であるKlf4と相互作用し、Tertの発現を制御します。この研究は、β-カテニンがテロメアの長さを制御することを通じて、幹細胞とがん性の間に新たな関連性を明らかにしました。
Takeoら(2013年)は、マウスの爪幹細胞(NSCs)が爪近位基質に存在し、ケラチン-14、ケラチン-17、KI67の高発現によって特徴づけられることを示しました。NSCの分化は趾の再生能力と直接関連しています。Wntに依存した分化を経た初期の爪前駆細胞は、切断後の爪の再生に必要であり、Wnt活性化が趾の再生にも寄与します。
Beronjaら(2013年)は、マウスを用いた皮膚発生とがん原性過形成に焦点を当てたゲノムワイドRNA干渉スクリーニングを行い、新たながん関連因子としてMllt6とβ-カテニンを同定しました。β-カテニンは、Wnt非依存的な細胞間接着を介して正常な表皮増殖の予期せぬアンタゴニストとして機能することが明らかにされました。
Kodeら(2014年)は、マウスの骨芽細胞におけるβ-カテニンの活性化変異が急性骨髄性白血病(AML)の発症につながることを示しました。β-カテニンはノッチリガンドJag1の発現を刺激し、造血幹細胞前駆細胞でのNotchシグナリングの活性化を引き起こします。Notchシグナリングの阻害はAMLの改善に寄与し、この病理におけるNotch経路の役割を証明します。
Descheneら(2014年)は、マウスの毛包幹細胞内でβ-カテニンが活性化すると、新しい毛髪の成長が誘発されることを示しました。この過程は毛髪再生に通常必要な間葉系のシグナルとは独立しています。
Diasら(2014年)は、マウスのD2型中棘ニューロンがβ-カテニンを介して抗不安作用と回復促進作用を媒介することを発見しました。β-カテニンはレジリエンスに関連するマイクロRNAの制御を介して、行動的な回復力の発達に重要な役割を果たします。
Benham-Pyleら(2015年)は、静止上皮細胞に機械的ひずみを加えると、YAP1とβ-カテニンの活性化が細胞周期の再突入を誘導することを発見しました。このプロセスはE-カドヘリンの細胞外ドメインの関与を必要とします。
Spadoniら(2015年)は、腸-血管バリア(GVB)の存在を評価し、そのサイズ排除特性とβ-カテニンシグナリングを介した血管透過性と細菌の侵入の制御機構を明らかにしました。
分子遺伝学
痙性片麻痺と視覚欠損を伴う神経発達障害
痙性片麻痺と視覚欠損を伴う神経発達障害に関する分子遺伝学的研究では、CTNNB1遺伝子の変異が重要な役割を果たしていることが示されています。CTNNB1遺伝子は、細胞の接着とシグナル伝達に関与するβ-カテニンタンパク質をコードしており、この遺伝子の変異は神経発達障害の原因となることがあります。
de Ligtら(2012年)は、痙性斜頸と視覚障害を伴う神経発達障害(NEDSDV; 615075)の3人の患者において、CTNNB1遺伝子にヘテロ接合性の機能喪失型変異を同定しました。この変異のうち2つは、患者で新規に生じた(de novo)ものであることが明らかにされました。
さらに、DECIPHERデータベースを利用したKharbandaら(2017年)の調査では、CTNNB1遺伝子の不活性化変異を有する11人の患者が同定されました。これらの変異は、さまざまな神経発達障害の原因となる可能性があります。
Liら(2017年)は、滲出性硝子体網膜症(EVR)と一致する眼の特徴、小頭症、発達遅延などを示した生後15ヵ月の男児において、CTNNB1遺伝子におけるde novoナンセンス変異(Q558X)のヘテロ接合を同定しました。
Panagiotouら(2017年)も、EVR、顔面異形、および発達遅滞を有する3歳の男児において、CTNNB1遺伝子の1bp挿入のヘテロ接合性を同定しました。
これらの研究は、CTNNB1遺伝子の変異が神経発達障害のスペクトラム内でさまざまな臨床的特徴を示す患者群において発生していることを示しており、β-カテニン経路の異常がこれらの疾患の発症に重要な役割を果たしている可能性があります。これらの発見は、神経発達障害の分子基盤の理解を深め、将来の治療戦略の開発に貢献することが期待されます。
滲出性硝子体網膜症7
Panagiotouら(2017年)の研究により、滲出性硝子体網膜症7(EVR7;617572)という遺伝性網膜疾患が、CTNNB1遺伝子の変異によって引き起こされることが示されました。CTNNB1遺伝子はβ-カテニンをコードしており、このタンパク質は細胞接着およびWntシグナル伝達経路において中心的な役割を果たします。この研究では、日本人の血縁関係のない2家系の罹患者から、CTNNB1遺伝子における2つの異なる変異が同定されました。一つはミスセンス変異R710C(116806.0024)、もう一つは切断変異(116806.0025)です。これらの変異はいずれもヘテロ接合性で発現していました。
β-カテニンは、細胞内外のシグナルを受け取り、遺伝子発現の調節に影響を与えることで細胞の運命を決定する重要な役割を担っています。そのため、CTNNB1遺伝子の変異は細胞機能に重大な影響を及ぼし、結果として網膜の発達や機能に異常を引き起こす可能性があります。滲出性硝子体網膜症は、網膜に異常な血管が形成されることを特徴とする疾患であり、視力低下や失明につながるリスクがあります。
この研究によって同定されたCTNNB1遺伝子の変異は、滲出性硝子体網膜症の病因解明に貢献し、将来的な治療法の開発に向けた新たな可能性を示唆しています。また、遺伝性網膜疾患の診断において、遺伝子検査の重要性を強調しています。
体細胞突然変異
Morinら(1997)、Ilyasら(1997)、Rubinfeldら(1997)、およびFearon(1997)の研究は、CTNNB1遺伝子の体細胞突然変異ががんの進行にどのように関与しているかを示しています。これらの研究を通じて、CTNNB1遺伝子における変異がβ-カテニンのアップレギュレーションや安定化につながり、がん細胞の異常な増殖や生存に寄与するメカニズムが解明されました。
Morinら(1997)は、CTNNB1遺伝子の体細胞突然変異がAPCを介したβ-カテニンのダウンレギュレーションに対して細胞を鈍感にし、大腸腫瘍形成において重要であることを示しました。
Ilyasら(1997)は、大腸癌細胞株の中にCTNNB1遺伝子の変異を持つものがあることを発見し、これらの変異がβ-カテニンと細胞接着タンパク質の複合体形成に影響を与える可能性があることを示しました。
Rubinfeldら(1997)は、メラノーマ細胞株におけるβ-カテニンの異常な蓄積がCTNNB1遺伝子の変異やAPC遺伝子の変異によって引き起こされることを発見し、これがメラノーマの進行に関与する可能性があることを示しました。
Fearon(1997)は、APC遺伝子とCTNNB1遺伝子の変異ががん細胞におけるβ-カテニンレベルの制御機構をどのように変化させるかを詳述し、これが細胞増殖の制御にどのように影響を及ぼすかを解説しました。
Chanら(1999年)による研究では、ヒト毛母腫の75%にCTNNB1の突然変異が見られ、これが毛母細胞の腫瘍形成の主要な原因であることが示唆されました。また、寒河江ら(1999年)やWrightら(1999年)の研究は、原発性卵巣癌や子宮内膜様卵巣癌においてもCTNNB1の変異が確認され、β-カテニンの細胞内蓄積と核への移行が関与していることが示されました。
特に、CTNNB1遺伝子の変異はβ-カテニンの安定化に関連しており、通常はGSK3βによるリン酸化とユビキチンを介した分解によって制御されています。これらの変異は、GSK3βのコンセンサス部位におけるセリン/スレオニン残基の変化を引き起こし、その結果、β-カテニンの分解が抑制され、細胞増殖シグナルが強化されます。
Kochら(1999年)による研究は、散発性肝芽腫においてもCTNNB1の変異が一般的であることを示しています。特に、これらの変異はβ-カテニンの分解を阻害し、転写活性の増加につながります。これは、β-カテニンが発生における成長促進シグナル伝達経路の中心的エフェクターであり、APCとともに細胞増殖と分化の調節に重要な役割を果たしていることを裏付けています。
APC遺伝子とCTNNB1遺伝子(β-カテニンをコード)に関する研究は、がん発生の分子メカニズムにおけるそれらの役割を浮き彫りにしました。APCは正常細胞でβ-カテニンのレベルを制御し、細胞増殖を抑制する役割を果たしています。APC遺伝子の不活性化やCTNNB1遺伝子の活性化変異は、β-カテニンの蓄積を引き起こし、腫瘍の形成に寄与することが明らかにされています。
特に、Blakerら(1999年)の研究は、肝芽腫の細胞質および核でβ-カテニンレベルが増加していることを示し、CTNNB1遺伝子のエクソン3に活性化変異が存在することを発見しました。この発見は、β-カテニンの蓄積が肝芽腫の発生に関与している可能性があることを示しています。
また、Legoixら(1999年)の研究は、肝細胞癌の一部でβ-カテニンの活性化変異が見られる一方で、APC変異は検出されなかったことを明らかにしました。さらに、β-カテニン変異の有無と特定の染色体セグメントの欠失との相関関係を示し、発がんには複数のメカニズムが関与している可能性があることを示唆しました。
Huangら(2000年)は、髄芽腫の一部でβ-カテニンのミスコーディング変異が存在し、特にコドン33に位置する変異が複数見られたことを発見しました。この結果は、β-カテニンシグナル伝達経路が髄芽腫の発生に関与していることを示しています。
Desmoid腫瘍や肺がん、悪性中皮腫など、他の腫瘍型においてもβ-カテニンやAPC遺伝子の変異が発見されています。これらの研究は、β-カテニンの蓄積やAPC遺伝子の不活性化が、さまざまな種類のがんにおいて共通の発がんメカニズムであることを示しています。
毛母腫におけるβ-カテニンの核発現は、Wnt/Ctnnb1/Tcf-Lef経路が毛包の正常なマトリックス細胞で活性化され、毛幹への分化を誘導することを示唆しています。さらに、β-カテニン変異が毛母腫の発生に関与していることが示されました。
これらの研究は、APC遺伝子の不活性化やCTNNB1遺伝子の活性化変異が多くのがん種の発生に重要な役割を果たしており、これらの遺伝子変異の検出はがん診断や治療の標的として有用であることを示しています。
動物モデル
Gatら(1998年)の研究では、表皮プロモーターで制御されたβ-カテニンの発現がマウスの毛髪形成に影響を及ぼし、新しい毛包の形成を引き起こすことが報告されました。これは、β-カテニンの安定化が毛髪形成の重要な調節因子であり、毛髪腫瘍の発生において異常なβ-カテニンの活性化が関与している可能性があることを示唆しています。
Haradaら(1999年)は、マウスでβ-カテニンの一部を標的欠失させた結果、Apcノックアウトマウスに似た腺腫性腸ポリープが生じることを見出し、β-カテニンが腸の発生と疾患に重要であることを示しました。
Huelskenら(2001年)は、マウスの表皮と毛包におけるβ-カテニンの条件付き突然変異が毛包形成を阻害し、毛髪サイクルに影響を与えることを報告しました。これは、β-カテニンが毛包の発生と維持に必須であることを示しています。
Saadi-Kheddouciら(2001年)は、β-カテニンのがん化型の過剰発現がマウスの腎臓に重度の多発性嚢胞病変を引き起こすことを発見し、β-カテニンが腎臓の発生と疾患に関与していることを示しました。
ChennとWalsh(2002年)は、β-カテニンのアミノ末端切断型の過剰発現が脳の肥大を引き起こし、これが前駆細胞集団の拡大によるものであることを示しました。これは、β-カテニンが哺乳類の脳の発達において重要な役割を果たしていることを示しています。
これらの研究は、β-カテニンが細胞の発生、組織の形成、そして特定の疾患の進行におけるキープレーヤーであることを強調しています。β-カテニンの活性化、安定化、およびその相互作用パートナーとの結合は、多くの生物学的プロセスを調節し、異常なβ-カテニンシグナリングは様々な病態に関与することが示されています。
Lickertら(2002)は、マウス胚において、β-カテニン遺伝子を条件付きで不活性化することで、中胚葉の形成には影響がないものの、結節の欠損、頭部と体幹のパターニングへの影響、ノトコルドや体節の形成不全を引き起こすことを発見しました。さらに、確定内胚葉でβ-カテニンを欠損させると、胚の前後軸に沿って複数の心臓が形成される異常が観察されました。
Soshnikovaら(2003)は、マウスにおいてWnt/β-カテニンシグナリングが頂端外胚葉隆起(AER)の形成と四肢の背腹軸パターニングに重要であることを明らかにしました。これらの過程は、Wnt/β-カテニンシグナリングとBMP受容体シグナリングの相互作用によって厳密に制御されています。
Xuら(2003)は、Ctnnb1を欠損したT細胞を有するマウスにおいて、脾臓T細胞数の大幅な減少とTCR刺激に対する反応の低下を観察しましたが、細胞死の亢進は見られませんでした。これは、β-カテニンがT細胞発生において重要な役割を果たしていることを示唆しています。
Dayら(2005年)は、異所性の正準Wntシグナリングが骨化を促進し、軟骨細胞形成を抑制することを発見しました。一方で、β-カテニンの遺伝的不活性化は軟骨細胞の異所性形成を引き起こし、骨芽細胞分化を阻害しました。
Hillら(2005年)は、マウスの間充織におけるβ-カテニンの条件的欠失が、骨芽細胞系の分化に必要であることを見出しました。β-カテニンが欠如すると、骨芽細胞前駆体は分化を阻害され、軟骨細胞に発達しました。
Glassら(2005)は、骨芽細胞を標的としたβ-カテニンの機能獲得または機能喪失変異を持つマウスが、それぞれ骨量の多いまたは少ない表現型を示すことを発見し、このプロセスが正統的Wntシグナリングによって制御されていることを示しました。
Francoら(2005)は、ヘリコバクター・ピロリ感染が胃の異形成と腺癌の発症を促進し、この過程でCtnnb(β-カテニン)の活性化が関与していることを示しました。これは、ヘリコバクター・ピロリ感染が胃癌リスクの増大に寄与する可能性のあるメカニズムを提供します。
これらの研究は、β-カテニンが胚発生、免疫応答、骨と軟骨の形成、さらにはがん発生に至るまで、生物学的プロセスの広範な範囲にわたって重要な役割を果たしていることを示しています。
Zamoraら(2007年)の研究では、マウスの前心膜でβ-カテニンを特異的に欠損させると冠動脈形成障害が起こりますが、静脈系と微小血管系は影響を受けませんでした。これは、心外膜の発達障害により、心筋への浸潤が鈍化し、心外膜由来の間葉系細胞から冠動脈平滑筋細胞への分化が不全になることを示しています。
プラナリアの再生能力に関するGurleyら(2008年)の研究では、β-カテニンのRNA干渉によって、尾の再生ではなく頭部が不適切に再生されることが明らかにされました。これはβ-カテニンがプラナリアの再生において前後性を決定する分子スイッチとして機能していることを示唆しています。
Liuら(2009年)は、マウスの生殖腺の発達におけるβ-カテニンの役割を調べ、β-カテニンが卵巣分化には必要であるが、精巣の発達には不要であることを示しました。β-カテニンの欠損は卵巣の発達障害を引き起こし、特定の遺伝子の発現パターンに影響を与えました。
Berthonら(2010年)による副腎皮質癌の研究では、β-カテニンが構成的に活性化されると副腎皮質での異形成が進行し、最終的には悪性特性を発現することが示されました。これは、β-カテニンが副腎皮質腫瘍の発生と進行に関与していることを示しています。
Tucciら(2014年)は、「バットフェイス」と呼ばれる変異体マウスを用いてβ-カテニンの変異が頭蓋顔面異常や脳形態学的変化を引き起こすことを示しました。これはβ-カテニンが頭部の形態形成と脳発達に重要な役割を果たしていることを示しています。
Dongら(2016年)は、自閉症スペクトラム障害(ASD)のモデルとしてパルバルブミン介在ニューロンでβ-カテニンを欠損させたマウスを研究しました。この変異マウスは不安症状の増大や社会的相互作用の障害などASDに似た行動を示し、β-カテニンの欠損が神経発達障害にどのように関与しているかの理解を深めました。
アレリックバリアント
.0001 大腸がん、体細胞性
CTNNB1、3-bp欠失、SER45DEL
全長APCを発現し、β-カテニンとTCF7L2による転写活性化の阻害を免れている2つの大腸癌(114500参照)細胞株において、Morinら(1997)はAPC癌抑制経路の下流の構成要素、すなわちCTNNB1遺伝子に変異を見出した。各腫瘍株は異なる変異を有していた:一方ではアミノ酸(ser45)を除去する3-bpの欠失、他方ではser33をtyr(116806.0002)に変化させるC-to-Aミスセンス変異であった。最初の患者のパラフィン包埋保存組織を分析した結果、この変異の体細胞性と培養前の原発腫瘍における存在が確認された。両変異とも、リン酸化によるβ-カテニンのダウンレギュレーションに関与しているセリンに影響を及ぼしていた。
.0002 大腸癌、体細胞性
毛母腫、体細胞、含む
CTNNB1、SER33TYR
116806.0001およびMorinら(1997)を参照。Ilyasら(1997)が大腸癌(114500参照)細胞株で発見した5つの点突然変異の1つは、CTNNB1遺伝子のエクソン3におけるCからAへのトランスバージョンによるser33からtyrへの突然変異であった。この変異はヘテロ接合型で存在した。
Chanら(1999年)は、毛母腫16例中2例(132600例)にこの変異を同定した。
.0003 肝芽腫、体細胞性
デスモイド腫瘍、体細胞、含む
CTNNB1, THR41ALA
6例の散発性肝芽腫(114550を参照)において、Kochら(1999)はCTNNB1遺伝子のコドン41にAからGへの転移を発見し、thr41からala(T41A)への置換をもたらした。(Iwaoら(1998)は散発性大腸癌におけるコドン41の変異を報告している)。T41A変異と肝芽腫を有する患者の年齢は4ヵ月から27ヵ月であった。
肝芽腫において、Blakerら(1999年)は、隣接する正常肝組織と比較して強い細胞質β-カテニン染色と腫瘍細胞核へのβ-カテニンの蓄積を示した。さらに、ある症例の腫瘍はコドン41がACC(thr)からGCC(ala)に変換するA-to-Gのヘテロ接合体であった。Legoixら(1999年)は肝細胞癌の3症例でこの同じ変異を発見した。これらの症例は小児例ではなく成人であった(平均年齢58歳;範囲27〜76歳)。98例の症例では、被験者の多くがアルコール依存症であった。
Shitohら(1999)は、散発性疾患の患者由来のデスモイド腫瘍組織内にCTNNB1遺伝子の体細胞性T41A変異を同定した(135290を参照)。
.0004 体細胞性肝芽腫
毛母腫、体性、含む
CTNNB1、ASP32TYR
2例の散発性肝芽腫(114550を参照)において、Kochら(1999)はコドン32がGACからTACに変化し、asp32からtyrに置換していることを発見した。患者の年齢は19ヵ月と30ヵ月であった。
Chanら(1999年)は、毛母腫16例中1例(132600例)にこの変異を同定した。
.0005 肝芽腫、体細胞性
CTNNB1, GLY34VAL (rs28931589)
散発性肝芽腫の3症例(114550を参照)において、Kochら(1999)は、コドン34がGGAからGTAに変化することにより、腫瘍がβ-カテニンのgly34からvalへの置換を有することを発見した。患者の年齢は10ヵ月から19ヵ月であった。
.0006 毛母腫、体性
CTNNB1, ASP32GLY
毛母腫16例中1例(132600例)において、Chanら(1999年)はCTNNB1遺伝子のAからGへの転移を発見し、β-カテニンのコドン32においてaspからglyへの置換をもたらした。
.0007 毛母腫、体細胞性
髄芽腫、体性、含む
CTNNB1, SER33PHE
調査した毛母腫16例中2例(132600例)において、Chanら(1999年)はCTNNB1遺伝子のC-T転移を同定し、その結果β-カテニンのコドン33(S33F)においてser-phe置換が生じた。
Huangら(2000年)は、散発性髄芽腫46例中3例(155255例)にCTNNB1遺伝子のS33F変異を同定した。
.0008 毛母腫、体細胞腫
CTNNB1, Gly34GLU
毛母腫16例中3例(132600例)において、Chanら(1999年)は、β-カテニンのコドン34におけるglyからgluへの置換をもたらすCTNNB1遺伝子のGからAへの転移を同定した。
.0009 毛母腫、体細胞性
CTNNB1, SER37CYS
調査された毛母腫16例中1例(132600例)において、Chanら(1999年)はCTNNB1遺伝子のCからGへの転座を同定し、その結果βカテニンのコドン37においてserからcysへの置換が生じた。
.0010 毛母腫、体細胞性
CTNNB1, SER37PHE
毛母腫16例中1例(132600例)において、Chanら(1999年)はCTNNB1遺伝子のC-T転移を同定し、β-カテニンのコドン37においてser-phe置換を生じた。
.0011 毛母腫、体細胞性
CTNNB1, THR41ILE
調査された毛母腫16例中1例(132600例)において、Chanら(1999年)はCTNNB1遺伝子のC-T転移を同定し、その結果β-カテニンのコドン41においてthr-ile置換が生じた。
.0012 卵巣がん、体細胞
CTNNB1, SER37CYS
Sagaeら(1999)が上皮性卵巣癌(167000)において検出したCTNNB1遺伝子のエクソン3における3つの変異の1つは、ser37-to-cys(S37C)ミスセンス変異であった。この腫瘍は子宮内膜様組織であった。
.0013 肝細胞がん、体細胞性
CTNNB1, SER45PHE
4例の肝細胞癌(114550)において、Legoixら(1999)はCTNNB1遺伝子のコドン45にTCT(ser)からTTT(phe)への変化を認めた。他の4症例ではser45からproへの変異がみられた(116806.0014)。
.0014 肝細胞がん、体細胞性
CTNNB1, SER45Pro
4例の肝細胞がん(114550)において、Legoixら(1999)はCTNNB1遺伝子のコドン45にTCT(ser)からCCT(pro)への変化を認めた。
.0015は116806.0007に移動した。
.0016 毛母腫、体細胞性
CTNNB1、ASP32TYR
毛母腫11例中3例において、Moreno-Buenoら(2001年)はCTNNB1遺伝子のエクソン3にヘテロ接合性のG-T転座を発見し、これによりasp32-tyr(D32Y)のアミノ酸変化が生じた。
.0017 痙性片麻痺と視覚障害を伴う神経発達障害
CTNNB1、4-bp欠失、NT1272
痙性斜頸と視覚障害を伴う神経発達障害(NEDSDV; 615075)を有する29歳の女性において、de Ligtら(2012)はCTNNB1遺伝子に4bpの欠失(1272_1275del)を同定し、フレームシフト(Ser425ThrfsTer11)を生じた。この変異は両親のどちらにも同定されなかった。著者らは、この患者はARFGEF2遺伝子にもヘテロ接合性のミスセンス変異(605371;R802Q)を有していることを指摘した。ARFGEF2遺伝子の変異は知的障害を引き起こすことが知られているが、常染色体劣性遺伝する。
.0018 痙性片麻痺および視覚障害を伴う神経発達障害
CTNNB1, ARG515TER
痙性斜頸と視覚欠損を伴う神経発達障害(NEDSDV; 615075)を持つ個体において、de Ligtら(2012年)はCTNNB1遺伝子のarg515-to-ter(R515X)のde novoヘテロ接合性ナンセンス変異を同定した。
.0019 痙性片麻痺と視覚障害を伴う神経発達障害
CTNNB1, GLN309TER
痙性斜頸と視覚欠損を伴う神経発達障害(NEDSDV; 615075)を有する個体において、de Ligtら(2012)はCTNNB1遺伝子のヘテロ接合性ナンセンス変異、gln309-to-ter(Q309X)を同定した。この変異は患者の母親には存在しなかったが、父親のDNAは検査に利用できなかった。
.0020痙性片麻痺と視覚障害を伴う神経発達障害
CTNNB1, 1-bp Dup, NT705
痙性斜頸と視覚障害を伴う神経発達障害(NEDSDV; 615075)の個体において、Tucciら(2014)は、フレームシフトと早期終止(Gly236ArgfsTer35)をもたらす、de novo heterozygous 1-bp duplication(705dup)を同定した。
.0021 神経発達障害(痙性両麻痺と視覚障害を伴う
CTNNB1, ARG535TER
痙性斜頸と視覚障害を伴う神経発達障害(NEDSDV; 615075)の血縁関係のない2人の患者において、Kharbandaら(2017年)は、CTNNB1遺伝子におけるde novo c.1603C-T転移のヘテロ接合性を同定し、arg535-to-ter(R535X)置換をもたらした。
.0022 痙性片麻痺と視覚障害を伴う神経発達障害
CTNNB1, GLN558TER
痙性斜頸と視覚障害を伴う神経発達障害(NEDSDV;615075)を有する15ヵ月の中国人男児において、Liら(2017年)は、CTNNB1遺伝子のエクソン11におけるde novo c.1672C-T転移のヘテロ接合性を同定し、gln558-to-ter(Q558X)置換をもたらした。
.0023 痙性片麻痺および視覚障害を伴う神経発達障害
CTNNB1, 1-bp ins, 1434c
痙性斜頸と視覚障害を伴う神経発達障害(NEDSDV; 615075)を有する3歳の中国人男児において、Panagiotouら(2017)は、CTNNB1遺伝子のエクソン9におけるde novo 1-bp挿入(c.1434_1435insC、NM_001904.3)のヘテロ接合性を同定し、早期終止コドン(Glu479ArgfsTer18)をもたらすと予測されるフレームシフトを引き起こした。この変異は罹患していない両親にもdbSNP、Exome Variant Server、ExACデータベースにも認められなかった。
.0024 滲出性硝子体網膜症 7
CTNNB1, ARG710CYS
滲出性硝子体網膜症(EVR7; 617572)を有する3世代の日本人家系(F410)の罹患メンバーにおいて、Panagiotouら(2017)はCTNNB1遺伝子のエクソン14におけるc.2128C-T転移(c.2128C-T, NM_001904.3)のヘテロ接合性を同定し、その結果、C末端ドメイン内の高度に保存された残基においてarg710からcys(R710C)への置換が生じた。この変異は発端者の9歳の罹患していない兄にもみられたが、dbSNP、Exomeバリアントサーバー、ExACデータベースでは発見されなかった。
.0025 滲出性硝子体網膜症 7
CTNNB1、16bp重複、NT2142
滲出性硝子体網膜症(EVR7; 617572)を有する日本人由来のハワイ系3世代家族(F258)の3人の罹患者において、Panagiotouら(2017)は16bp重複のヘテロ接合性を同定した(c.2142 2157dupTAGCTATCGTTCTTTT, NM_001904.3)は、CTNNB1遺伝子のエクソン15にフレームシフトを引き起こし、C末端ドメイン内に早期終止コドン(H720X)をもたらした。この変異は家族内で疾患と分離し、dbSNP、エクソームバリアントサーバー、ExACデータベースでは発見されなかった。







