目次

ALG3-CDGは、ALG3遺伝子の機能喪失型変異によって引き起こされる、世界でわずか約43例しか報告されていない極めて希少な先天性代謝異常症です。小胞体でのN-グリコシル化が根本から障害されることで、乳児期早期から重度の神経発達障害・難治性てんかん・多関節拘縮が複合的に出現する、深刻な多臓器発現型疾患です。
Q. ALG3-CDGとはどのような疾患ですか?まず結論だけ知りたいです
A. ALG3遺伝子の機能喪失型変異による常染色体劣性遺伝の先天性グリコシル化異常症(CDG)です。小胞体でのN-グリコシル化経路の中心的酵素が欠損することで、重度の神経発達障害・難治性てんかん・多関節拘縮・視覚障害が乳児期から複合的に現れます。現在根本的治療法はなく、対症療法が中心です。
- ➤疾患の定義 → OMIM #601110(CDG1D)、Orphanet ORPHA:79321、世界での報告例約43例
- ➤分子メカニズム → 小胞体内腔でのMan5→Man6変換が停止し、Man5中間体が蓄積・小胞体ストレス慢性化
- ➤主な症状 → 重度精神運動発達遅滞・難治性てんかん(ウエスト症候群等)・多関節拘縮・視神経萎縮
- ➤診断 → トランスフェリン等電点電気泳動(Type Iパターン)+質量分析+次世代シーケンシング
- ➤治療・展望 → 現在は対症療法中心。遺伝子治療・フェロトーシス誘導療法など基礎研究が急速に進展
1. ALG3-CDGとは:疾患の定義と命名の変遷
ALG3-CDG(OMIM #601110)は、ALG3遺伝子(Asparagine-Linked Glycosylation 3)のホモ接合体または複合ヘテロ接合体の機能喪失型変異によって引き起こされる、常染色体劣性遺伝形式をとる先天性代謝異常症です。世界中で報告されている患者数は約43例という極めて希少な疾患であり、日本では「先天性糖鎖修飾異常症」として小児慢性特定疾病の対象に含まれます。
💡 用語解説:CDG(先天性グリコシル化異常症)とは
CDG(Congenital Disorders of Glycosylation)とは、細胞内でタンパク質や脂質に糖鎖を付加する「グリコシル化」の過程に関わる酵素の遺伝的欠損によって引き起こされる先天性疾患群の総称です。現在160種類以上のサブタイプが報告されており、ALG3-CDGはそのうちの一つです。グリコシル化はタンパク質の正常な折りたたみ・細胞への輸送・免疫応答に不可欠であるため、その障害は全身の臓器に重大な影響を及ぼします。
この疾患の命名には歴史的な変遷があります。かつては「糖質欠損糖タンパク質症候群IV型(CDGS4)」や「先天性グリコシル化異常症 Type Id(CDG-Id / CDG1D)」と呼ばれてきましたが、近年は原因遺伝子を明記する国際標準の命名法に従い「ALG3-CDG」と呼称されます。
国際希少疾患データベースOrphanetには「ORPHA:79321」として登録されています。ALG3遺伝子はヒトゲノムの第3染色体長腕(3q27.1)に位置し、進化的に高度に保存されたハウスキーピング遺伝子として、哺乳類から酵母・紅藻にいたるまで相同遺伝子が存在します。
💡 用語解説:常染色体劣性遺伝とは
常染色体(性染色体以外の染色体)上の遺伝子について、両方のコピー(父親由来・母親由来)に変異が存在するときにのみ症状が現れる遺伝形式です。両親はそれぞれ1コピーずつ変異を持つ「保因者(キャリア)」であり、通常は無症状です。保因者の夫婦から生まれる子どもが同疾患を発症する確率は理論上25%です。ALG3-CDGはこの遺伝形式をとるため、患者の両親は通常健康です。
2. ALG3遺伝子とN-グリコシル化の分子メカニズム
ALG3-CDGの病態を理解するには、細胞内でタンパク質に糖鎖を付加する「N-グリコシル化」という生化学プロセスの仕組みを把握することが不可欠です。このメカニズムこそが、ALG3遺伝子の欠損が全身の臓器に多大な影響をもたらす根本的な理由です。
💡 用語解説:N-結合型グリコシル化(N-グリコシル化)とは
タンパク質のアスパラギン(N)残基に糖鎖(オリゴ糖)が結合する翻訳後修飾のことです。タンパク質の正しい三次元構造の形成・細胞内での輸送・細胞膜上での安定的な発現・免疫認識を決定する最も重要な修飾の一つです。ほぼすべての分泌タンパク質や膜タンパク質がN-グリコシル化を受けており、その障害は広範な臓器機能不全に直結します。
LLO生合成経路:フェーズIとフェーズII
N-グリコシル化の出発点は、脂質結合型オリゴ糖(LLO:Lipid-Linked Oligosaccharide)と呼ばれる糖鎖前駆体の合成です。この過程は小胞体(ER)膜を挟んで2つのフェーズに厳格に区別されます。
💡 用語解説:小胞体(ER)とドリコールリン酸(Dol-P)
小胞体(ER:Endoplasmic Reticulum)は細胞内に網目状に広がる膜系オルガネラで、タンパク質の合成・折りたたみ・修飾の主要な場です。ドリコールリン酸(Dol-P)は小胞体膜に組み込まれた脂質分子で、LLO合成の「土台(足場)」として機能します。糖鎖はこのDol-Pに順番に積み上げられ、最終的にタンパク質へ一括転移されます。
フェーズI(細胞質側)では、ALG7をはじめとする一連の酵素が順番に作用し、Dol-Pに2つのGlcNAc(N-アセチルグルコサミン)と5つのマンノースが付加されてMan5GlcNAc2-PP-Dolという中間体が完成します。この中間体は、RFT1タンパク質(フリッパーゼ)の作用によって小胞体膜を「フリップ(反転)」し、小胞体内腔側に移行します。
フェーズII(小胞体内腔側)の最初のステップを担うのがALG3酵素です。小胞体膜に組み込まれ、活性部位を内腔に向けたALG3は、ドリコールリン酸マンノース(Dol-P-Man)を糖ドナーとして利用し、フリップされたMan5GlcNAc2-PP-DolのC-6位マンノース残基にα-1,3結合で6番目のマンノースを付加します。この反応によってMan6GlcNAc2-PP-Dolが生成されます。
このALG3による反応は、後続のALG9酵素への基質を提供する律速段階として機能します。ALG3が欠損すると、Man5GlcNAc2-PP-Dol中間体が小胞体内腔に蓄積し、完全な14糖前駆体(Glc3Man9GlcNAc2-PP-Dol)が形成できなくなります。その結果、オリゴ糖転移酵素(OST複合体)によるタンパク質への糖鎖転移効率が著しく低下し、細胞全体で広範な低グリコシル化(ハイポグリコシル化)が生じます。
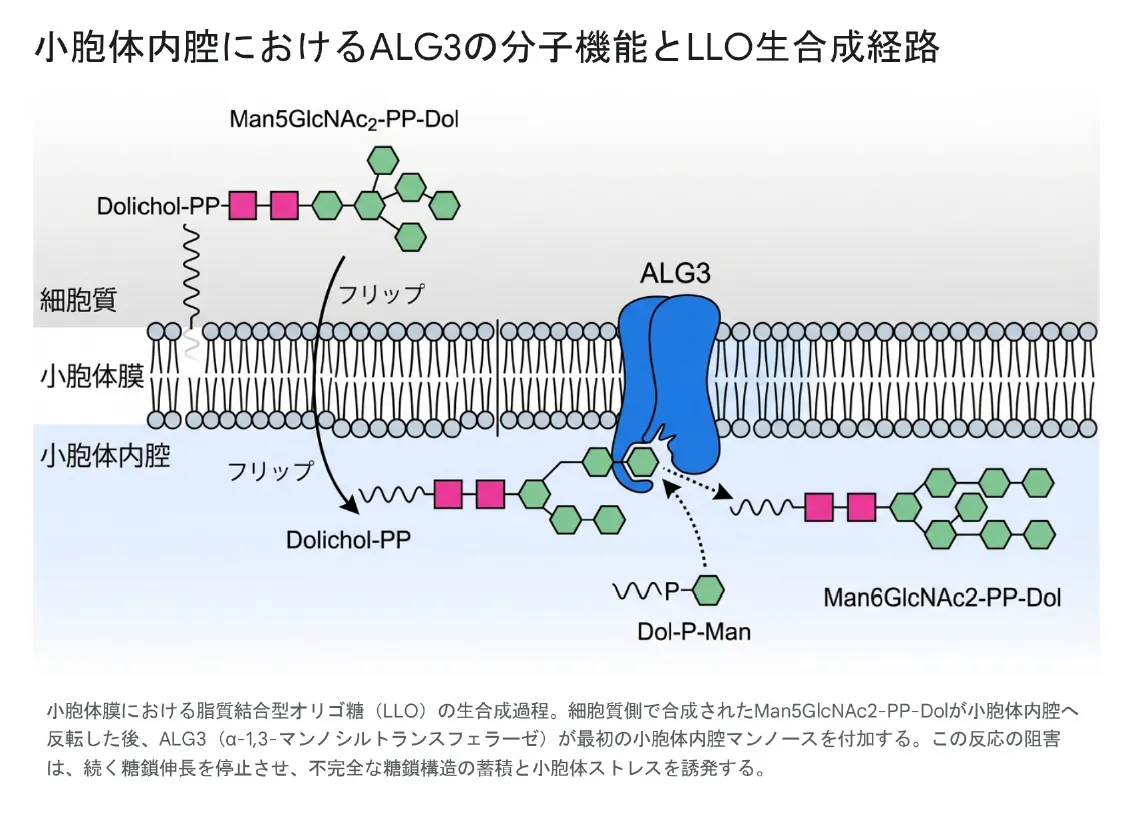
小胞体膜におけるLLO生合成経路。細胞質側で合成されたMan5GlcNAc2-PP-DolがRFT1によって小胞体内腔へ反転した後、ALG3(α-1,3-マンノシルトランスフェラーゼ)が最初の内腔側マンノースを付加する。ALG3が欠損するとこの反応が停止し、不完全な糖鎖構造の蓄積と小胞体ストレスが誘発される。
小胞体ストレスの慢性化とUPR(異常タンパク質応答)
ALG3欠損の病態の核心は、単なる「糖鎖の未完成」という静的な構造欠陥にとどまりません。細胞全体の品質管理機構が慢性的に破綻する点にあります。蓄積したMan5中間体により新生タンパク質の正しいフォールディング(折りたたみ)が妨げられ、これが強力な小胞体ストレス(ERストレス)を惹起します。
💡 用語解説:UPR(Unfolded Protein Response/異常タンパク質応答)とは
小胞体内で折りたたみに失敗したタンパク質が蓄積すると、細胞はストレスを感知して一連の適応応答を起動します。これをUPRといいます。正常時は一時的な保護機構として機能しますが、ALG3-CDGでは解決できないストレスが恒常的にUPRを活性化し続けます。具体的には、ERストレスセンサーIRE1-α経路が持続的に活性化し、転写因子XBP1 mRNAの特異的スプライシングが誘導されます。ALG3-CDG患者細胞では、スプライシング型XBP1の発現が健常細胞の2倍に過剰発現することが確認されています。この慢性的なUPR活性化と、それに伴う過剰なタンパク質分解(ERAD亢進)が、脳・神経系を中心とした重篤な臓器障害の根本的な分子病態です。
3. 主な症状と全身性臨床像
ALG3-CDGは乳児期早期から発症する多臓器発現型の重篤な代謝異常症です。中枢神経系を主体とした深刻な発達障害に加え、骨格系・眼科的異常・肝機能障害が複合的に発現します。中国の同胞例では、羊水検査を通じて胎生期における複合的なグリコシル化障害が検出され、胚性致死・胎生致死をとる重症例の存在も明らかになっています。
🧠 中枢神経・神経学的異常
- 重度精神運動発達遅滞・知的能力障害
- 顕著な筋緊張低下(フロッピーインファント)
- ヒプスアリスミアを伴うてんかん(ウエスト症候群等)
- 進行性の脳萎縮・小脳萎縮
- 脳梁形成不全
👁️ 眼科的異常
- 斜視・近視
- 視神経萎縮
- 虹彩欠損(コロボーマ)
- 網膜ジストロフィー
- 角膜混濁・進行性の視力喪失
🦴 筋骨格・結合組織
- 多発性関節拘縮症(AMC)
- 脊柱側弯症(神経筋性)
- 内反足
- 低骨量・点状軟骨異形成症
🫀 消化器・肝臓・血液系
- 哺乳困難・成長障害
- 肝酵素上昇・肝腫大
- 血液凝固異常(出血傾向)
- 反復性感染症
- 心拡張・心肥大・心膜胸水
💡 用語解説:多発性関節拘縮症(AMC:Arthrogryposis Multiplex Congenita)
先天的に複数の関節が固まった(拘縮した)状態で生まれてくる疾患です。胎児期に関節の動きが著しく制限されることで起こり、筋肉の発達不全や結合組織の異常が背景にあります。ALG3-CDGでは、広範な糖タンパク質の低グリコシル化が結合組織の異常発達を招くことで、AMCを発症すると考えられています。
💡 用語解説:ヒプスアリスミア(Hypsarrhythmia)とウエスト症候群
ヒプスアリスミアとは、脳波上に現れる高振幅の混沌とした不規則な棘徐波パターンのことです。これを伴うてんかんの一形態がウエスト症候群(点頭てんかん)で、生後3〜12か月に発症することが多く、シリーズ形成する突然の頭部前屈発作(点頭発作)を特徴とします。ALG3-CDGでは、このウエスト症候群を含む難治性てんかんが高頻度に合併し、精神運動発達に深刻な影響を与えます。
特徴的な顔貌異常(ディスモルフィズム)
ALG3-CDGでは、眼球隔離症(眼の間が広い)・幅広く平坦な鼻梁・低く傾斜した額・分厚く大きな耳・薄い唇・小顎症または突出した下顎・高口蓋など、特徴的で広範な顔面形態異常が報告されています。こうした顔貌は診断の手がかりとなりますが、類似した顔貌を示す他疾患との鑑別が必要です。また、ホルモン異常(女児の無思春期)・難聴・神経管閉鎖不全を伴う重症例も存在します。

4. 鑑別診断:他のCDGおよび関連疾患との違い
ALG3-CDGの診断において最大の課題の一つは、同じ「Type Iパターン」を示す他のCDGサブタイプや、類似した神経症状を呈する他の先天性疾患との鑑別です。特に血清トランスフェリン検査のみでは同一グループ内の疾患を区別できないため、精密な分子診断が必須となります。
PMM2-CDG との鑑別
重要:CDGの中で最多を占める(全体の約70%)のがPMM2-CDGです。同じく「Type Iパターン」を示し、小脳萎縮・精神運動発達遅滞を呈するため初期に混同されやすい疾患です。
鑑別のポイント:原因遺伝子がPMM2(ゴルジ体のタンパク質マンノシル化経路に関与)であり、多関節拘縮やコロボーマの合併はPMM2-CDGでは稀。NGSによる遺伝子検査で確定します。
他のALG-CDG(ALG6・ALG8・ALG9等)との鑑別
注意点:ALG3と同じLLO生合成経路に関わるALG6、ALG8、ALG9等のCDGも同様の「Type Iパターン」を呈し、神経発達異常を共通して示します。
鑑別のポイント:特異的な糖鎖質量分析プロファイルの違い(ALG3-CDGではHex3-4HexNAc2の異常蓄積)と、遺伝子パネルまたはWES検査が確定に必要です。
RFT1-CDG(CDG-In)との鑑別
かつて文献で「CDG-In」とALG3-CDGが混同される記載があったため、整理が必要です。RFT1-CDGはフリッパーゼRFT1の欠損による別の独立した疾患です。
鑑別のポイント:RFT1-CDGではDol-PP-GlcNAc2Man5の反転(フリップ)が障害されるため、蓄積する中間体の種類が異なり、遺伝子検査(RFT1 vs ALG3)で明確に区別されます。
先天性筋疾患・てんかん性脳症との鑑別
筋緊張低下・難治性てんかん・発達遅滞という症状の組み合わせは、先天性ミオパチーや他のてんかん性脳症(CDKL5欠損症、Ohtahara症候群等)とも重なります。
鑑別のポイント:血液凝固糖タンパク質(アンチトロンビンIII等)の低下・肝酵素上昇・多関節拘縮の組み合わせがある場合は、TfIEFスクリーニングを優先的に実施することが推奨されます。
5. 診断・遺伝子検査のアプローチ
ALG3-CDGの診断は、生化学的スクリーニング → 精密質量分析 → 遺伝子検査という統合的なアルゴリズムによって確定されます。
ステップ1:トランスフェリン等電点電気泳動(TfIEF)によるスクリーニング
💡 用語解説:トランスフェリン等電点電気泳動(TfIEF)とは
血液中の鉄輸送タンパク質であるトランスフェリンは2か所のN-グリコシル化部位を持ちます。正常ではシアル酸が4つ結合した「テトラシアロトランスフェリン」が主成分ですが、CDGによる低グリコシル化が生じると、シアル酸数の少ないアイソフォーム(ジシアロ・アシアロ)が増加し、電気泳動パターンがカソード側にシフトします。これを「Type Iパターン」と呼び、ALG3-CDGを含む多くのLLO組み立て欠陥型CDGで検出されます。比較的簡便に実施でき、CDGのスクリーニング第一選択です。
ステップ2:LC-ESI-MS(液体クロマトグラフィー質量分析)による糖鎖プロファイリング
TfIEFではCDGの大分類(Type I / Type II)は判別できますが、どの遺伝子が原因かを特定することはできません。近年、RapiFluor標識を用いたLC-ESI-MS(液体クロマトグラフィー・エレクトロスプレーイオン化質量分析)の導入により、分子レベルの精密診断が可能となりました。
スロバキアの3歳男児の症例(ALG3遺伝子のc.165C>T〔p.Gly55=〕及びc.1060C>T〔p.Arg354Cys〕の複合ヘテロ接合体変異)のLC-ESI-MS解析では、正常な高次マンノース構造(Hex6-8HexNAc2)の相対存在量が著しく減少し、ALG3機能不全を直接反映する低マンノース構造(Hex3HexNAc2、Hex4HexNAc2)が血清中に異常蓄積していることが明確に証明されました。これらの異常糖鎖構造はALG3-CDGに対する高感度・高特異度の疾患バイオマーカーとして応用が期待されています。
ステップ3:次世代シーケンシング(NGS)による確定診断
💡 用語解説:全エクソームシーケンシング(WES)と遺伝子パネル検査
WES(Whole Exome Sequencing)は、遺伝子のタンパク質コード領域(エクソン)全体を網羅的に解析する次世代シーケンス手法です。ALG3-CDGのように希少で症例報告が少ない疾患では、一度の検査で多くの候補遺伝子を評価できるWESまたはCDG専門の遺伝子パネル検査が推奨されます。なお、常染色体劣性遺伝疾患の場合、両親を含めたトリオ解析を行うことでde novo変異との区別や保因者確認が可能です。遺伝子検査の詳細はこちら
確定診断においては、遺伝子検査で同定されたALG3変異が機能喪失型(ナンセンス変異・フレームシフト変異・スプライス部位変異)か、または機能欠失が確認されたミスセンス変異であることを確認します。必要に応じて患者の皮膚線維芽細胞を用いた酵素活性測定や、細胞レベルでのN-グリコシル化の生化学的解析によって変異の病原性を証明します。
血清TfIEF(Type Iパターン確認)→ LC-ESI-MSによるN-グリカンプロファイリング(Hex3-4HexNAc2蓄積の確認)→ CDG遺伝子パネルまたはWES(ALG3病原性変異の同定)→ 必要に応じて機能解析(皮膚線維芽細胞の酵素活性測定)
6. 治療と長期管理
現時点では、ALG3-CDGに対する根本的な疾患修飾治療(遺伝子治療・酵素補充療法等)は確立されていません。治療は多臓器に及ぶ合併症に対する包括的な対症療法が中心となります。小児科・神経科・眼科・整形外科・肝臓科・臨床遺伝科が連携した集学的チーム医療体制の構築が不可欠です。
🧠 てんかんへの対応
ウエスト症候群を含む難治性てんかんに対しては、ACTHアナログ・ビガバトリン・抗てんかん薬(バルプロ酸・クロナゼパム等)を用います。発作のコントロールは神経発達の保護に直結するため、神経科専門医による定期的な脳波評価が必要です。
🦴 整形外科的管理
多発性関節拘縮症・内反足・脊柱側弯症に対しては、理学療法・装具療法・必要に応じた外科的矯正手術を実施します。早期からのリハビリテーション開始が関節可動域の維持と生活の質(QOL)改善に重要です。
👁️ 眼科的管理
コロボーマ・視神経萎縮・網膜ジストロフィーに対する定期的な眼科評価と、残存視力の最大化を目指した早期介入が重要です。斜視への手術療法および眼鏡矯正を適切なタイミングで実施します。
🫀 栄養・肝臓・血液管理
哺乳困難・成長障害に対しては経管栄養の導入を含む栄養サポートが必要です。肝酵素の定期モニタリング・凝固系の評価(アンチトロンビンIII・第XI因子等)を定期的に実施し、出血リスクを管理します。
将来の根本治療に向けた研究の最前線
ALG3-CDGに対する将来の疾患修飾治療として、研究の最前線では以下の2つのアプローチが有望視されています。
①遺伝子治療・バイパス経路の薬理学的活性化:欠損したALG3酵素機能を補完する遺伝子治療、または蓄積するMan5中間体を処理するバイパス経路の活性化が研究されています。LC-MSを用いた分子診断技術の確立が、こうした標的治療の開発基盤を整えています。
②腫瘍学領域からのスピンオフ:「免疫原性フェロトーシス」誘導研究:悪性腫瘍研究においてALG3阻害が「免疫原性フェロトーシス」を誘導するメカニズムが解明されつつあります(がん細胞においてALG3過剰発現は治療抵抗性と関連)。この成果はALG3の生化学的経路を深く解明するものであり、CDG-Id研究にも新たな知見をもたらす可能性があります。ただし、がん細胞での「ALG3阻害戦略」とALG3-CDGの「ALG3欠損状態」とは全く別の文脈であり、臨床応用には慎重な検討が必要です。
7. 遺伝カウンセリング
ALG3-CDGの確定診断後、家族への丁寧な遺伝カウンセリングが必要です。常染色体劣性遺伝疾患であるALG3-CDGの遺伝カウンセリングでは、以下の内容を中心に話し合います。
- ➤再発リスクの説明:両親はともに保因者(ヘテロ接合体)であるため、次子が同疾患を発症する確率は理論上25%です。この確率は毎回の妊娠で独立して生じます。
- ➤保因者検査:家族の変異が同定された場合、同胞・近親者への保因者検査を提案できます。保因者は通常無症状ですが、将来の家族計画に重要な情報となります。
- ➤出生前診断の選択肢:次子を望む場合、絨毛検査・羊水検査による出生前遺伝子診断が選択肢として存在します。既知の両親の病原性変異が確認されている場合、確実な診断が可能です。体外受精と組み合わせた着床前遺伝学的検査(PGT)も選択肢として検討できます。
- ➤予後情報の提供:現時点では根本的治療法がなく、重度の神経学的障害が持続する例が多いことを伝えながら、対症療法によるQOL改善の可能性と、継続的な医療・リハビリテーション支援の重要性を説明します。
- ➤患者支援情報:国際的なCDG患者ネットワーク(CDG Care、FCDGC等)や国内の先天性代謝異常症支援団体の情報を提供し、他の家族とのつながりを支援します。
8. よくある誤解
誤解①「CDG-IdとCDG-Inは同じ疾患だ」
一部のデータベースで「CDG-In」と関連付けられる記述が散見されますが、これは過去の文献の混同です。CDG-Id(ALG3-CDG)はALG3欠損、CDG-In(RFT1-CDG)はフリッパーゼRFT1欠損による独立した別疾患です。分子機序も原因遺伝子も異なります。
誤解②「トランスフェリン検査で確定診断できる」
TfIEFはCDGのスクリーニング検査であり、Type Iパターンを示しても多くのCDGサブタイプ(PMM2-CDG、他のALG-CDG等)が該当します。確定診断には必ず遺伝子検査(NGS)が必要であり、さらに質量分析による糖鎖プロファイリングが診断の精度を高めます。
誤解③「代謝異常症だから食事療法で治る」
PKUやGalactosemiaのように食事療法が有効なCDGサブタイプ(MPI-CDG等)も存在しますが、ALG3-CDGは現在根本的治療が存在せず、食事療法による改善は見込めません。対症療法と集学的管理が治療の中心です。
誤解④「両親が健康なのだから遺伝疾患ではない」
ALG3-CDGは常染色体劣性遺伝のため、両親はそれぞれ1コピーずつ変異を持つ「保因者」であり通常は完全に健康です。「両親が健康だから遺伝性ではない」という思い込みが、診断の遅れにつながることがあります。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少遺伝性疾患・先天性代謝異常症について
ALG3-CDGをはじめとする希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] NCBI Gene. ALG3 alpha-1,3-mannosyltransferase [Homo sapiens (human)]. Gene ID: 10195. [NCBI Gene]
- [2] GeneCards. ALG3 Gene – GeneCards | ALG3 Protein | ALG3 Antibody. [GeneCards]
- [3] CDG Hub. ALG3-CDG (CDG-Id). [CDG Hub]
- [4] Orphanet. ALG3-CDG. ORPHA:79321. [Orphanet]
- [5] NIH Genetic and Rare Diseases Information Center. ALG3-congenital disorder of glycosylation. [NIH GARD]
- [6] Deficient glycan extension and endoplasmic reticulum stresses in ALG3-CDG. PMC. 2024. [PMC11251843]
- [7] Insights into ALG3-CDG: A case study combining glycan profiling and genetic analysis. PMC. 2025. [PMC12517101]
- [8] Freeze HH, et al. Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview. GeneReviews. NCBI Bookshelf. [NCBI Bookshelf]
- [9] N-Linked Protein Glycosylation in the Endoplasmic Reticulum. PMC. 2013. [PMC3721281]
- [10] Foundation for Congenital Disorders of Glycosylation (FCDGC). ALG3-CDG. [FCDGC]
- [11] Congenital Disorders of Glycosylation from a Neurological Perspective. PMC. 2021. [PMC7827962]
- [12] The glycosyltransferase ALG3 is an AKT substrate that regulates protein N-glycosylation. PMC. 2025. [PMC12451169]






