目次

ACTB遺伝子は、私たちの体のほぼすべての細胞でβ(ベータ)アクチンというタンパク質をつくるとても基本的で重要な遺伝子です。第7染色体(7p22.1)に位置し、細胞の形を保ったり、動いたり、分裂したりするときに欠かせません。近年は、生まれつきの病気(バライツァー・ウィンター症候群など)やがんの進行・転移にも深く関わることがわかってきています。
Q. ACTB遺伝子とはどんな遺伝子ですか?まず結論だけ知りたいです
A. 第7染色体(7p22.1)にあり、βアクチンというタンパク質をつくる遺伝子です。βアクチンは細胞の骨組み(細胞骨格)の主役で、細胞の形・運動・分裂を支えます。変異の場所や種類によって、神経・血液・聴覚の病気を引き起こすことが知られており、がんの進行や転移にも関わる重要な遺伝子です。
- ➤遺伝子の基本 → 第7染色体7p22.1、βアクチンをつくる中核遺伝子
- ➤細胞での働き → 形・運動・分裂・神経・免疫など多彩な役割
- ➤翻訳の仕組み → ZBP1という運搬役による精緻な「場所決め」翻訳
- ➤関連する疾患 → バライツァー・ウィンター症候群、ジストニア・難聴症候群、症候性血小板減少症
- ➤がんや検査 → 多くのがんで発現上昇、検査では「偽遺伝子」への注意が必要
1. ACTB遺伝子とは:基本情報
ACTBは「Actin Beta(アクチン・ベータ)」の略で、第7染色体の短い腕の先端近く(7p22.1)に位置する遺伝子です。この遺伝子からつくられるβアクチンは、進化の過程で魚や鳥から人まで、ほとんど形を変えずに受け継がれてきたほど大切なタンパク質で、ヒトの細胞では最も豊富に存在するタンパク質の一つです。
💡 用語解説:アクチンとは
アクチンは細胞の中で「骨組み」や「線路」のような役割を果たすタンパク質です。脊椎動物には大きく3種類(α・β・γ)があり、α型は筋肉で力を出す主役、β型とγ型は筋肉以外のあらゆる細胞で、形を保ったり物を運んだりする働きをしています。本記事の主役であるβアクチンは「細胞質アクチン1」とも呼ばれます。
細胞の中でβアクチンは2つの形を行き来しています。バラバラの粒のような「Gアクチン(球状アクチン)」と、それらが鎖のようにつながった「Fアクチン(線維状アクチン)」です。この2つの形を行き来する仕組みが、細胞の運動・分裂・物質輸送・遺伝子発現の調整など、生命のあらゆる場面の原動力になっています。
2. 遺伝子の構造とβアクチンの作られ方
ACTB遺伝子の本体は第7染色体上で約3,500塩基対の領域に収まっており、ここから1,812塩基のメッセンジャーRNA(mRNA)がつくられます。興味深いのは、ACTBが2種類の少しずつ違うmRNA(オルタナティブ転写産物)を生み出すことです。この多様性が、後ほど説明する細胞内での「翻訳の場所決め」の柔軟性につながっています。
💡 用語解説:mRNA(メッセンジャーRNA)と翻訳
遺伝子(DNA)の情報をタンパク質に変換する「中継役」がmRNAです。DNAの情報を写し取ったmRNAは、細胞の中でリボソームという翻訳工場に運ばれ、アミノ酸が順番につながれてタンパク質になります。この「DNA → mRNA → タンパク質」の流れを、生物学ではセントラルドグマと呼びます。
出来上がったβアクチンには、状況に応じてさまざまな「化学的な飾り付け(翻訳後修飾)」が施されます。代表的なのはアルギニル化と呼ばれる修飾で、これによって細胞が動くときの最前線(葉状仮足)でのβアクチンの働きが微調整されます。また、ウイルス感染時にはISG化という別の修飾を受け、免疫の働きを助けることもわかってきました。
3. 細胞内での主な働き
βアクチンはかつて「ただの骨組み」とみなされていましたが、現在では細胞のあらゆる活動の中心に関わるダイナミックなタンパク質と理解されています。主な役割を整理してみましょう。
🧱 形と接着の維持
細胞の形を保ち、隣の細胞としっかり結合するための土台になります。腸の表面のような上皮細胞の頂部と底部の区別を保つのもβアクチンの仕事です。
🏃 細胞の運動
線維芽細胞や免疫細胞などが動くとき、進行方向の最前線(葉状仮足)でβアクチンが急速に重合し、推進力を生み出します。傷の修復や免疫応答に欠かせません。
🧠 神経のしなやかさ
神経細胞のシナプス(情報の受け渡し場所)の動的な変化や、血液脳関門の維持にも重要です。記憶や学習を支える分子としても注目されています。
🛡️ 免疫の補佐役
細胞質だけでなくミトコンドリアの中にもβアクチンが存在し、ウイルス感染時の自然免疫スイッチ(IRF3)を安定させる、新しい役割が次々に発見されています。
このように、βアクチンは細胞の形を保つだけのタンパク質ではなく、運動・接着・神経・免疫・分裂といった生命のあらゆる場面で主役級の働きをしているのです。
4. βアクチンが「必要な場所で必要な時に」つくられる仕組み
細胞が動いたり神経が枝を伸ばしたりするとき、βアクチンは細胞のあちこちで均等につくられるのではなく、必要な場所でピンポイントに合成される必要があります。この「場所決め翻訳(局所翻訳)」の鍵を握るのが、ACTB mRNAの末尾にある特殊な配列と、それを認識するタンパク質です。
💡 用語解説:ジップコードとZBP1
ACTB mRNAの末端(3’非翻訳領域)には「ジップコード」と呼ばれる郵便番号のような短い配列があります。ZBP1(Zipcode-Binding Protein 1、別名IGF2BP1)というタンパク質がこの配列に結合することで、mRNAは目的地まで「宛先付きの荷物」のように運ばれます。鳥類から哺乳類まで共通して受け継がれた、進化的に古い精密な仕組みです。
ZBP1がmRNAに結合すると、mRNAは約180度のループ状に折り曲げられ、リボソーム(翻訳工場)がアクセスできない状態になります。つまりこの段階では、βアクチンタンパク質はまだつくられません。「翻訳されない状態」のまま、mRNAは細胞内の線路(微小管)に沿って目的地まで運ばれていくのです。
目的地(細胞が動こうとしている最前線など)に到着すると、Srcキナーゼという酵素がZBP1にリン酸という化学修飾をつけます。するとZBP1がmRNAから外れ、ようやくリボソームが結合してβアクチンの翻訳が始まります。こうして「必要な場所で、必要な時だけ」βアクチンが供給される仕組みが完成しています。
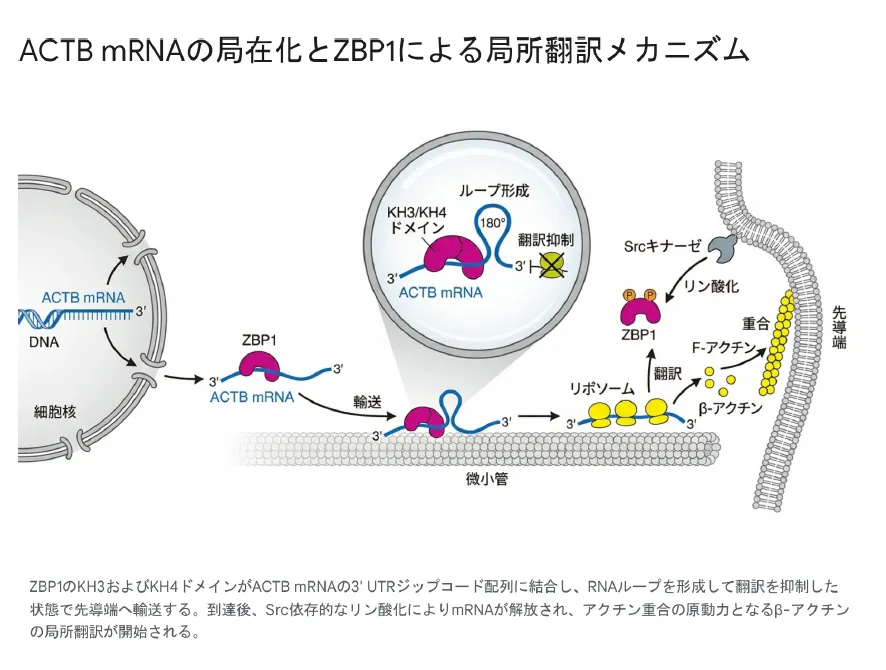
ZBP1のKH3およびKH4ドメインがACTB mRNAの3’UTRジップコード配列に結合し、RNAループを形成して翻訳を抑制した状態で先導端へ輸送する。到達後、Src依存的なリン酸化によりmRNAが解放され、アクチン重合の原動力となるβアクチンの局所翻訳が開始される。
🧬
①核内で転写
DNAから ACTB mRNAが合成される
🔗
②ZBP1が結合
3’末端のジップコードを認識
🔁
③ループ形成
mRNAが180度折れて翻訳ストップ
🚂
④輸送
微小管に沿って先導端へ移動
⚡
⑤リン酸化
SrcキナーゼがZBP1を解除
🏭
⑥局所翻訳
先導端でβアクチンが合成される
ZBP1により翻訳を抑制された状態で運ばれ、目的地で初めてβアクチンが合成される仕組み
5. βアクチンとγアクチンの違い
非筋細胞で働くアクチンには「β型(ACTB由来)」と「γ型(ACTG1由来)」の2種類があります。両者のアミノ酸配列は99%同じで、違いはたった4つのアミノ酸のみ。それなのに、マウスでACTBを完全に欠損させると胎児の段階で命を落とす(胎生致死)のに対し、ACTG1を欠損させても生まれてくることができます。
長年、これは「βアクチンというタンパク質そのものが生命に必須だから」と説明されてきました。しかし2018年に発表された画期的な研究が、この常識を覆します。研究者たちは、ACTB遺伝子の調節領域(プロモーターやジップコード)はそのまま残しつつ、つくられるアミノ酸配列だけをγ型に変えたハイブリッドマウスを作出しました。
💡 驚きの結果
体内からβアクチンタンパク質が完全に失われたにもかかわらず、このマウスは正常に発生し、生まれ、成長し、運動機能も野生型とほぼ同じでした。つまり生命の維持に必須だったのは「βアクチンタンパク質そのもの」ではなく、ACTB遺伝子の塩基配列に埋め込まれた調節機構(プロモーターやジップコード)だったのです。
ただし、このハイブリッドマウスには一つだけ問題が現れました。加齢に伴い、進行性の高音域難聴を発症したのです。これは、内耳の感覚細胞(有毛細胞)の中の極細の毛(不動毛・ステレオシリア)を維持するためには、βアクチンタンパク質固有の性質が必要であることを示しています。同じように見えるβとγにも、完全には置き換えがきかない、それぞれ固有の役割があることがわかってきました。
6. ACTBの変異が引き起こす生まれつきの病気
ACTB遺伝子に生まれつき変化(変異)があると、全身のさまざまな細胞で細胞骨格の働きが乱れ、複数の臓器に影響が及ぶ病気を引き起こします。これらは非筋性アクチノパシー(ノンマッスル・アクチノパシー)と総称されています。特徴的なのは、変異の場所や種類によって、まったく異なる病気が起きることです。
💡 用語解説:機能獲得型と機能喪失型、新生突然変異
機能獲得型(Gain-of-Function)変異とは、タンパク質に新たな(多くは有害な)働きが加わったり、構造変化で正常タンパク質を邪魔したりするタイプです。
機能喪失型(Loss-of-Function)変異はタンパク質の働きが失われるタイプ。
新生突然変異(de novo変異)とは、両親には変異がなく、お子さんで初めて生じた変化のことです。
① バライツァー・ウィンター脳前頭顔面症候群(BWCFF)
最もよく知られているのがバライツァー・ウィンター脳前頭顔面症候群(BWCFF)です。ACTBに「機能獲得型」のミスセンス変異(特にp.Arg196Hisが代表的)が生じることで起こります。常染色体顕性(優性)遺伝の形式をとりますが、ほとんどは新生突然変異によるもので、両親には変異がないことが多いです。
主な特徴は、両眼が離れて見える「両眼開離(ハイパーテロリズム)」・先天性の眼瞼下垂・前頭部の縫合線の盛り上がり(三角頭蓋)といった特徴的な顔つき、軽度から重度の知的障害、てんかん、そして大脳の前頭部優位の厚脳回(脳のしわが少なく分厚い)などです。多くの方で肩甲帯筋の萎縮、感音難聴、関節の動かしにくさなども見られます。
② ジストニア・難聴症候群(DDS)
ACTBにp.Arg183Trpという特定の変異が起こると、BWCFFとはまったく違う症状が出る「ジストニア・難聴症候群」を発症します。主な症状は薬で抑えにくい重いジストニア(体の一部が意図せず緊張・ねじれる動き)と若年発症の感音難聴で、発達の遅れや背骨の曲がり(側弯症)を伴うこともあります。同じ遺伝子の変異でも、変化したアミノ酸が違うだけで、影響を受ける場所がまったく変わるという、遺伝学の不思議さがよくわかる例です。
③ 症候性血小板減少症(THC8)
一方、ACTB遺伝子のエクソン5・6(遺伝子の3’末端側)に「機能喪失型」の変異が起こると、まったく別の病気「巨大血小板を伴う症候性血小板減少症(THC8)」が現れます。骨髄で血小板がつくられる最終段階の細胞分裂が物理的に止まってしまい、大きすぎる血小板が、数も少ない状態で末梢の血液に放出されます。多くの場合、明らかな自然出血はありませんが、軽度の発達の遅れや小頭症、白血球の増加、繰り返す感染、光線過敏などを伴うことがあります。
④ 体細胞モザイク変異による皮膚疾患
ACTBの変異は、生まれる前の細胞分裂の途中で体の一部の細胞だけに生じることもあります。これを体細胞モザイク変異と呼び、この場合は全身ではなく、変異を持った細胞から派生した組織にだけ症状が現れます。代表的なのがベッカー母斑症候群と先天性平滑筋過誤腫で、片側性の色素沈着・多毛・平滑筋の過剰増殖といった皮膚病変として現れ、半側肥大を伴うこともあります。
📋 ACTB変異が引き起こす主なアクチノパシーの比較
| 症候群名 | 変異のタイプ | 病態の仕組み | 主な特徴 |
|---|---|---|---|
| バライツァー・ウィンター症候群 (BWCFF) |
ミスセンス変異 機能獲得型 (例:p.Arg196His) |
細胞骨格機能の異常により神経細胞の移動障害が起こる | 特徴的な顔貌・知的障害・前頭優位の厚脳回・難聴 |
| ジストニア・難聴症候群 (DDS) |
特定ミスセンス変異 p.Arg183Trp |
中枢神経・有毛細胞への局所的影響 | 薬剤抵抗性ジストニア・若年発症難聴・側弯症 |
| 症候性血小板減少症 (ACTB-AST/THC8) |
エクソン5・6の変異 機能喪失型 |
巨核球から血小板への成熟過程が阻害される | 巨大血小板・血小板減少・小頭症・反復感染 |
同じACTB遺伝子であっても、変異の場所と種類によって全く異なる病気が起こる「多面発現性」の代表例です。

7. がんとACTB:腫瘍学における役割
ACTBは健康な細胞でも非常に多く発現していますが、多くのがんで発現がさらに上昇することが、大規模ながん遺伝子解析(TCGAなど)でわかってきました。肝細胞がん、頭頸部扁平上皮がん、肺腺がん、胆管がん、食道がん、神経膠芽腫、腎臓がん、子宮頸がん、白血病、膵がんなどでACTBの過剰発現が報告され、これが患者さんの予後と関係することも示されています。
EMT(上皮間葉移行)と転移の主役の一人
がん細胞がもとの場所から離れて広がっていくとき、しっかり接着した上皮細胞から、動き回る間葉系の細胞へと姿を変えるEMT(上皮間葉移行)と呼ばれるプロセスが起こります。このときに必要なのが、細胞骨格の劇的な作り変えで、その中心にいるのがβアクチンです。
💡 用語解説:インバドポディア(浸潤突起)
悪性度の高いがん細胞は、表面に「インバドポディア」と呼ばれるアクチンに富んだ突起をつくります。この突起はマトリックスメタロプロテアーゼという酵素を集めて、周囲の組織を分解し、がん細胞が新たな場所に侵入する「道」をつくる役割を果たします。トリプルネガティブ乳がんなどの浸潤性が強いがんで特に活発です。
ACTBが他の遺伝子と「融合」して発がんを引き起こす
ACTBの活性化されたプロモーター(遺伝子のスイッチ部分)が、染色体の組み換えによりがん関連遺伝子のスイッチを乗っ取って強く活性化させてしまう「融合遺伝子」のケースも知られています。代表例は以下の2つです。
ACTB-GLI1融合
第7染色体と第12染色体が組み換わるt(7;12)転座によって生じ、「t(7;12)転座を伴う周皮腫」という特殊な腫瘍を引き起こします。ACTBの強いプロモーターがGLI1(がん遺伝子)のスイッチを乗っ取り、異常な活性化を生みます。
ACTB-FOSB融合
非常に稀な軟部腫瘍「偽筋原性血管内皮腫」の約半数で検出される融合遺伝子です。臨床的には他のサブタイプより単発の病変として現れやすい傾向があり、診断や治療方針を決める重要な手がかりとなります。
8. 検査・研究での利用と意外な落とし穴
ACTBは、遺伝子発現量を測定する基礎研究の現場で、長年にわたって「いつでもどこでも一定量つくられている前提の物差し」として使われてきました。これをハウスキーピング遺伝子(HKG)と呼びます。実際、2010〜2015年に発表された遺伝子発現研究の38%がACTBを「物差し」として使用していたという報告もあります。
しかし現在では、この使い方には2つの深刻な落とし穴があることがわかっています。
落とし穴①:ヒトゲノムに64個もの「偽遺伝子」が存在する
💡 用語解説:偽遺伝子(プソイドジーン)
偽遺伝子とは、本物の遺伝子とそっくりな配列を持ちながら、機能を失った「化石のような配列」のことです。ACTBにはヒトゲノム上に64個もの偽遺伝子が存在し、本物のmRNAと配列がよく似ています。検査でDNAが少しでも混入すると、本物と偽物を区別できずに測定値が最大6.5倍も過大評価されることがあります。
落とし穴②:がんや低酸素ストレスでACTBは大きく変動する
前のセクションで見たように、ACTBはがん組織で発現が大きく上昇します。「いつも一定」という前提が成り立たない状況で、「一定の物差し」として使うと、本当に変化させたい遺伝子の真の変動が見えなくなってしまいます。このため現在ではACTBは、正式ながん関連遺伝子のリファレンスからは外されつつあります。
🛠️ MIQEガイドラインに準拠した正確な発現解析のステップ
STEP 1
候補遺伝子を複数選ぶ
ACTB1個に頼らず、PPIA・UBC・HNRNPL・PCBP1など2〜3個以上を候補にする
STEP 2
アルゴリズムで安定性を検証
geNorm・NormFinder・BestKeeperなどのソフトで安定性スコアを評価
STEP 3
幾何平均で正規化
最も安定した複数遺伝子の幾何平均を使い、エクソン境界をまたぐ特異的プライマーで増幅
単一遺伝子(ACTBなど)に依存しない、再現性の高いデータを得るための国際ガイドライン
最新の研究では、甲状腺乳頭がんではSYMPK、汎がん解析ではHNRNPL・PCBP1・RER1などが、ACTBに代わる新しいリファレンス遺伝子として提案されています。「ACTBだから安心」という時代は終わり、研究対象と条件ごとに最適な物差しを選び直すパラダイムシフトが進んでいます。
9. 臨床遺伝専門医からのメッセージ
ACTB遺伝子は、ミネルバクリニックのインペリアルプラン(154遺伝子218疾患を対象とする出生前単一遺伝子パネル)の対象遺伝子の一つとして含まれています。バライツァー・ウィンター症候群をはじめ、出生前に把握できる場合があれば、新生児期からの集学的なケア計画につながることもあります。
ただし、ACTBに関連する病気は症状の幅が非常に広く、新生突然変異がほとんどです。家族歴がないからといって絶対に起こらないわけではなく、また検査結果が陽性だったとしても、その表現型は一人ひとり異なります。検査の意義や限界、結果が出た後の選択肢については、検査の前から遺伝カウンセリングで十分にお話しすることが大切です。
よくある質問(FAQ)
🏥 ACTB遺伝子・出生前診断について相談したい方へ
ACTB関連疾患を含む単一遺伝子疾患の出生前検査や、
結果解釈のご相談は、臨床遺伝専門医が在籍するミネルバクリニックへお気軽にどうぞ。
関連記事
参考文献
- [1] NCBI Gene. ACTB actin beta [Homo sapiens]. Gene ID: 60. [NCBI Gene 60]
- [2] UniProt. ACTB – Actin, cytoplasmic 1 (Human). UniProtKB P60709. [UniProt P60709]
- [3] Verloes A, et al. Baraitser-Winter Cerebrofrontofacial Syndrome. GeneReviews®. [NCBI Bookshelf NBK327153]
- [4] MedlinePlus Genetics. ACTB gene. National Library of Medicine. [MedlinePlus]
- [5] Chao JA, et al. ZBP1 recognition of β-actin zipcode induces RNA looping. Genes Dev. 2010;24(2):148-158. [PMC2807350]
- [6] Vedula P, et al. Essential nucleotide- and protein-dependent functions of Actb/β-actin. PNAS. 2018;115(50):E11541-E11550. [PNAS]
- [7] Latham SL, et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat Commun. 2018;9(1):4250. [PubMed 30315159]
- [8] Gu Y, et al. A pan-cancer analysis of the prognostic and immunological role of β-actin (ACTB) in human cancers. Bioengineered. 2021. [PMC8806805]
- [9] Antonescu CR, et al. A Distinct Malignant Epithelioid Neoplasm With GLI1 Gene Rearrangements (ACTB-GLI1 fusion). Am J Surg Pathol. 2018. [PubMed 29309307]
- [10] Sun Y, et al. Pseudogenes as Weaknesses of ACTB and GAPDH Used as Reference Genes in RT-PCR. PLoS One. 2012;7(8):e41659. [PMC3425558]
- [11] OMIM. ACTB actin, beta. #102630. Johns Hopkins University. [OMIM 102630]






