目次

10番染色体異常の全貌:トリソミー・モノソミーの症状と最新治療
📍 クイックナビゲーション
ヒトのゲノムは、生命の設計図として機能する複雑な分子ネットワークを形成しています。その中で10番染色体は、全体を構成するDNAの約4%〜4.5%を占める非常に重要な構造体です。
最新の細胞遺伝学的および分子生物学的なマッピングにより、10番染色体上には約800〜1200個のタンパク質コード遺伝子が配置されていることが明らかになっています。これらの遺伝子群は、胎生期における初期発生、心血管系の構築、中枢・末梢神経系の分化、免疫細胞(特にT細胞)の成熟など、生命維持の根源的な役割を担っています。
10番染色体異常の全体像
10番染色体における数的異常(染色体全体の過剰)や構造異常(一部の欠失や重複)は、遺伝子のコピー数変異(CNVs)を引き起こします。これにより「遺伝子量」の深刻な不均衡が生じ、単一の臓器にとどまらず、身体の複数の器官系にわたる重篤な先天的形態異常や発達遅滞を特徴とする多様な症候群を惹起します。
- ➤完全トリソミー10: 大半は胎児期に自然流産に至るが、モザイク型の場合は生存し得る
- ➤10p異常(短腕): HDR症候群(GATA3遺伝子)やDiGeorge様症候群など
- ➤10q異常(長腕): 10q26欠失症候群など、重度の神経発達障害(EBF3遺伝子)の関与
- ➤最新の医療動向: マイクロアレイ検査による精密診断と、将来の遺伝子治療の展望
1. 10番染色体の異常とは?遺伝子型と表現型の相関
過去数十年にわたり、これらの異常は主にGバンド分染法などの古典的な細胞遺伝学的検査によって大まかに分類されてきました。しかし、近年の染色体マイクロアレイ検査(CMA)や次世代シーケンシング(NGS)の劇的な進歩により、染色体の切断点(ブレイクポイント)が塩基対レベルで正確に特定されるようになりました。
この技術的ブレイクスルーにより、特定の微小な遺伝子領域の欠失や重複が、どのような臨床的表現型を引き起こすのかという「遺伝子型-表現型相関(Genotype-Phenotype correlation)」の解明が急速に進んでいます。例えば、10番染色体短腕(10p)のGATA3遺伝子や、長腕(10q)のEBF3遺伝子などが、特定の症候群の病態生理の中核であることが証明されつつあります。
💡 【用語解説】コピー数変異(CNVs)と遺伝子量
通常、染色体は両親から1本ずつ受け継ぎ、合計2本(2コピー)で機能します。コピー数変異(CNVs)とは、DNAの一部が欠失して1コピーになったり、重複して3コピー以上になったりする状態です。設計図である遺伝子のコピー数が増減すると、作られるタンパク質の量も変動し、この遺伝子量(Gene dosage)の不均衡が様々な疾患の根本原因となります。
2. 完全トリソミー10およびモザイクトリソミーの病態と予後
10番染色体全体が過剰になる状態には、すべての細胞が影響を受ける「完全トリソミー」と、正常細胞と異常細胞が混在する「モザイクトリソミー」があります。
2.1 完全トリソミー10(Full Trisomy 10)の致死性
完全トリソミー10は、すべての細胞において10番染色体が3本(合計47本)存在する、極めて重篤な異数性の状態です。親の生殖細胞形成時の染色体不分離によって自然発生的に引き起こされ、母体年齢が35歳を超えるとリスクが有意に上昇します。
ゲノム全体の遺伝子量が過剰になるため、胚の正常な発生プログラムは致命的に破綻します。その結果、完全トリソミー10を伴う妊娠の大部分は生命維持と適合せず、胎児期早期に自然流産に至るのが一般的です。極めて稀に出生したケースでも、重度の心不全や腎不全など多岐にわたる深刻な合併症を抱えており、新生児期・乳児期早期の死亡が最も可能性の高い転帰となります。
💡 【用語解説】胎盤限局性モザイク(CPM)とは
NIPT(非侵襲的出生前検査)でトリソミー10陽性となった場合、それが必ずしも胎児の完全トリソミーを意味するわけではありません。「胎盤限局性モザイク(CPM)」と言って、胎盤には異常細胞が存在するものの、胎児本体は正常な核型(46本)を維持しているケースが少なくありません。この場合、羊水検査による確定診断が極めて重要になります。
2.2 モザイクトリソミー10の臨床的プロファイル
受精後の細胞分裂エラーによって生じるモザイクトリソミー10は、体内に正常細胞が混在するため、完全トリソミーと比較して生児として出生する確率が高まります。しかし、生存した患児は依然として複雑で重篤な多臓器合併症に直面します。
- 消化器系の形態異常が顕著で、最も頻度が高いのは鎖肛(Anal atresia)です。
- 筋骨格系への影響も破壊的であり、重度の側弯症、関節の不可逆的な拘縮、合指症・多指症などが観察されます。
- 最も深刻な予後因子として、極めて重度な精神運動発達の遅滞が確認されており、自立歩行や言語獲得は著しく制限されます。
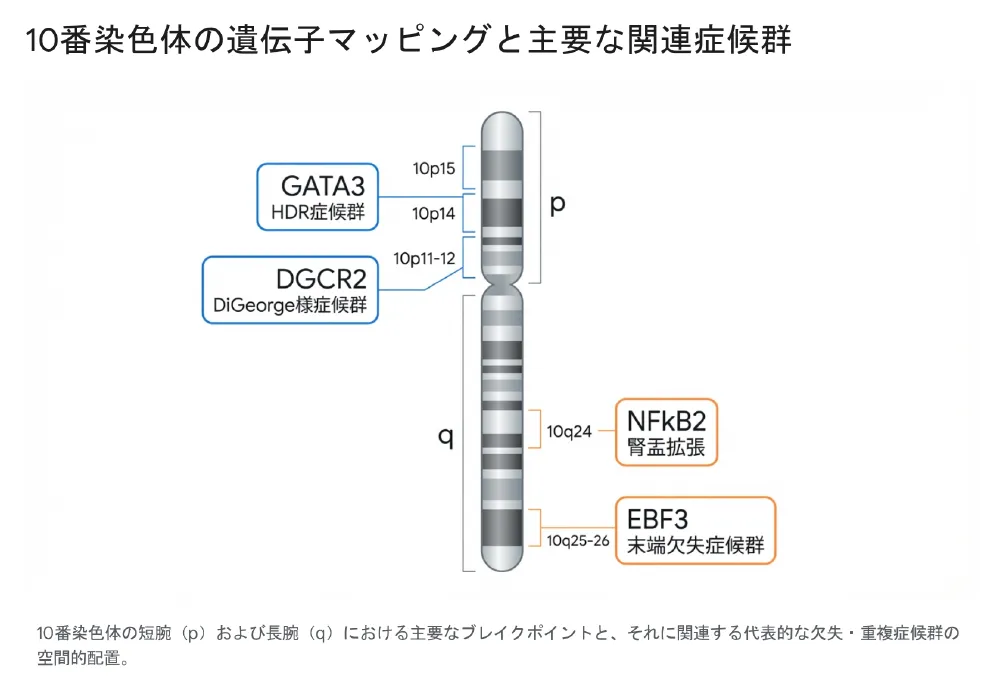
図:10番染色体の遺伝子マッピングと主要な関連症候群の空間的配置
3. 10番染色体短腕(10p)の部分的構造異常
染色体の一部分のみが重複または欠失する構造異常は、完全トリソミーよりも長期間生存するケースが多く、欠失・重複した特定の遺伝子群に由来する特異的な症候群を形成します。
3.1 部分トリソミー10p(重複症候群)
部分トリソミー10pは、短腕の全部または一部のセグメントが重複する稀少な染色体異常です。この発生メカニズムの大部分は突然変異ではなく、表現型が正常な親のいずれかが均衡型転座や腕間逆位といった構造的再編成を保因していることに起因します。
特徴的な症状として、頭蓋骨が前後に長くなる長頭症、異常に突出した前額部、広い鼻根部、口唇口蓋裂などが挙げられます。内臓器官では下肢の内反足が高頻度で発生するほか、腎臓における嚢胞性変化が特徴的な指標となります。運動・認知能力の発達は著しく遅延し、生涯にわたる支援が必須です。
3.2 部分モノソミー10p(欠失症候群)の病態生理
10pの一部が喪失する部分モノソミーは、欠失するDNAセグメントの物理的な大きさ(メガベース単位)とブレイクポイントの位置に依存して非常に多様な臨床像を呈します。近年の分子遺伝学的解析により、以下の明確な症候群に細分化されています。
また、この領域のより広範な欠失として10p14-p13欠失症候群も知られており、隣接する遺伝子群の欠落範囲によって発達遅滞などの症状や重症度が複雑に変化することが分かっています。
■ HDR症候群(Barakat症候群)と10p14欠失(GATA3遺伝子)
10p14領域に含まれるGATA3遺伝子の機能喪失は、HDR症候群の直接的な原因となります。GATA3は初期発生における必須の転写因子であり、これが失われる(ハプロ不全)ことで以下の3症状が発症します。
- H – 副甲状腺機能低下症:血中カルシウム濃度が低下し、生命を脅かす痙攣発作(テタニー)を誘発します。
- D – 難聴:両側性の感音性難聴として発症し、高音域の聴取が困難になります。
- R – 腎疾患:腎臓の低形成や嚢胞形成、進行性の慢性腎臓病(CKD)として現れます。
【最新の薬理学的アプローチ】
低カルシウム血症の管理には、従来ビタミンDとカルシウム製剤が用いられますが、過剰投与は腎結石の危険があります。近年、抗真菌薬であるフルコナゾール(Fluconazole)がビタミンDの分解酵素を強力に阻害し、カルシウム吸収効率を飛躍的に向上させる画期的な副次的効果を持つことが示唆されています。
■ DiGeorge様症候群と10p11-10p12欠失(DGCR2領域)
より動原体に近い近位欠失領域には、DGCR2(DiGeorgeクリティカルリージョン2)が存在します。この領域が失われると、22q11.2欠失(DiGeorge症候群)と驚くほど類似した症状が現れます。
- 心室中隔欠損症などの重度な先天性心疾患
- 胸腺発達障害による重篤な免疫不全と繰り返す呼吸器感染症
- 口蓋裂や鼻咽腔閉鎖不全(特有の鼻声や嚥下時の逆流の原因)
💡 【用語解説】ハプロ不全とは
通常、遺伝子は父親由来と母親由来の2つのコピーが揃って十分な機能を発揮します。そのうちの片方が欠失したり変異したりして機能しなくなり、残った1つのコピーだけでは必要なタンパク質量を賄いきれずに症状が出てしまう状態をハプロ不全(Haploinsufficiency)と呼びます。GATA3や後述するEBF3の異常は、まさにこのハプロ不全が原因で重篤な症状を引き起こします。
4. 10番染色体長腕(10q)の部分的構造異常
長腕(q腕)は物理的に長大であるため、重複や欠失が発生したブレイクポイントが「近位」か「遠位(末端)」かによって、関与する遺伝子群が異なり表現型が劇的に変化します。
4.1 部分トリソミー10q(重複症候群)
近位重複(10q11〜10q22): 小頭症、深く落ち窪んだ小さな眼、小さく後退した下顎などの頭蓋顔面異形が見られます。身体成長遺伝子が過剰発現することの逆説的な結果として、極めて著しい低身長(成人で138〜148cm程度)を示します。また、半数以上で重度の股関節脱臼が発生します。
遠位重複(10q23/24〜10qter): より重篤な多臓器異常を特徴とします。特に重要なのが10q24バンドに位置するNFkB2遺伝子の関与で、この重複による過剰発現は、腎盂拡張や水腎症の発生メカニズムと強く関連しています。
4.2 10q26欠失症候群(部分モノソミー10q)とEBF3遺伝子
10q末端(10q25〜10q26)の欠失は、明確な臨床的疾患単位を確立しています。鳥の嘴のように曲がった鼻、小頭症、両眼開離といった特徴的な顔貌に加え、男児においては小陰茎や停留精巣などの泌尿生殖器系の異常が非常に高い確率で発生します。
患者を生涯にわたって最も強く苦しめるのは、複雑かつ重篤な神経行動学的な障害です。発語の開始や運動スキルの獲得が大幅に遅延し、小児期後期からは極端な注意力欠如・多動性障害(ADHD)、深刻な衝動性、自閉症スペクトラム特有の行動が顕在化します。
近年の全エクソームシーケンシング(WES)等の網羅的解析により、この領域内に存在するEBF3遺伝子のハプロ不全が、中枢神経機能不全という複雑な神経行動学的プロファイルの中核的な役割を果たしていることが明らかになりました。
5. 10pおよび10q部分異常における臨床表現型の比較表
以下の表は、10番染色体短腕および長腕の構造異常における主要な臨床症状を比較したものです。各症候群に特徴的な表現型(例:10q26欠失におけるADHDや、10p重複における嚢胞腎など)が鑑別のポイントとなります。
| カテゴリー | 10p重複 (Partial Trisomy 10p) |
10q重複 (Partial Trisomy 10q) |
10q26欠失 (Partial Monosomy 10q) |
|---|---|---|---|
| 頭蓋顔面 (Craniofacial) |
・長頭、前頭部突出 ・大泉門・縫合の開大 ・広い鼻根 ・口唇口蓋裂 |
・小頭症、前頭部突出 ・平坦で厚い耳輪・後方回転耳 ・奥まった小さな目・斜視 ・内眼角贅皮、虹彩欠損、網膜異形成 ・上向きの鼻、弓状の口 ・高口蓋・口蓋裂、小顎症 |
・突出した/鷲鼻、広い鼻筋 ・変形した低位耳 ・薄い上唇、小顎症 ・小頭症 ・両眼開離、斜視 ・短く翼状の首 |
| 骨格 (Skeletal) |
・内反足 ・長く細い四肢 ・多指症・合指症 |
・低身長 ・側弯症 ・屈指症 |
・成長障害 ・側弯症 ・関節可動域制限 ・内反足 |
| 内臓・器官 (Visceral) |
・嚢胞腎 ・鎖肛 |
・心疾患 ・停留精巣 ・腎盂拡張・水腎症 |
・外性器異常(停留精巣、小陰茎など) ・腎・尿路異常 ・心疾患 |
| 神経・発達・行動 (Neuro/Behavioral) |
・筋緊張低下 ・けいれん |
・認知機能障害(学習支援が必要) ・発達遅滞(運動など) ・筋緊張低下 |
・軽度〜中等度の知的障害 ・発達遅滞(発語・運動) ・筋緊張低下 ・ADHD、衝動性制御困難、自閉症様行動 |
6. 染色体10異常の先進的診断法と医学的管理戦略
10番染色体の異常を正確に同定し、個々の表現型に応じた予後予測を行うためには、細胞遺伝学と分子遺伝学を融合させた多層的なアプローチを要します。
6.1 ゲノム医療に基づく多層的診断アプローチ
特異的な多発奇形や原因不明の発達遅滞が認められた場合、最初のステップとしてGバンド分染法による古典的な核型分析が行われます。しかし、顕微鏡下で検出できない微小欠失(Microdeletion)の特定には、全ゲノムのコピー数変異を高解像度でスキャンする染色体マイクロアレイ検査(CMA)が標準的な第一線の精密診断ツールとなります。ミネルバクリニックでも、NGS遺伝子検査パネルなど、最新の分子遺伝学的手法を用いた精密な診断を提供しています。
💡 【用語解説】顕性(優性)遺伝と潜性(劣性)遺伝
従来の「優性」「劣性」という言葉は、遺伝子の優劣と誤解されやすいため、現在では「顕性(けんせい)」「潜性(せんせい)」と表現します。
・常染色体顕性(優性)遺伝: ペアになる遺伝子のうち、片方に変異があるだけで症状が「顕れる」遺伝形式。HDR症候群などが該当します。
・常染色体潜性(劣性)遺伝: 両方の遺伝子に変異が揃った時にのみ症状が発現し、片方だけの場合は「潜む」遺伝形式。
6.2 集学的医療ケア(Multidisciplinary Care)の展開
現在、失われた染色体を細胞レベルで丸ごと補充するような根治的治療法は存在しません。したがって、医学的管理の基本戦略は、患者の全身に及ぶ多様な症状に対して、各専門科が連携して包括的に介入する「集学的管理」となります。
- 内分泌・代謝管理: 小児内分泌科による血清カルシウム等の厳密なモニタリングと、活性型ビタミンD等の処方調整(HDR症候群など)。
- 心血管・泌尿器外科的介入: 小児循環器・泌尿器科による生後早期の心エコー評価や修復手術、停留精巣の固定術など。
- 神経行動・発達支援: 児童精神科・小児神経科によるADHDや自閉症様症状への向精神薬投与と、PT・OT・STによる包括的療育プログラムの実行。
7. 最新の研究動向と将来の治療的パラダイムシフト
染色体異常に対する医療は長らく対症療法の枠を出ませんでしたが、分子生物学と遺伝子工学の爆発的な進歩により、希少遺伝疾患に対する「根本的・分子的介入」という新たなパラダイムが現実のものとなりつつあります。
2024年末、神経伝達物質の合成が阻害される超希少疾患に対し、正常な遺伝子を組み込んだ改変アデノ随伴ウイルス(AAV2)ベクターを脳に直接注入する初の遺伝子治療薬(Kebilidi)がFDAに承認されました。これは不治とされてきた遺伝的脳機能障害に対する直接的な治療が可能であることを証明した歴史的なマイルストーンです。
10番染色体異常のように複数の遺伝子が欠失する症候群であっても、GATA3やEBF3のような「マスターレギュレーター遺伝子」が次々と同定されています。現在、CRISPRを用いたゲノム編集や、エピジェネティックな発現制御によって細胞レベルで機能を修正する次世代の遺伝子治療アプローチが精力的に研究されています。
また、デューク大学などで進行中の画期的な臨床試験では、自己またはドナーの「臍帯血(UCB)」に由来する細胞を髄腔内に直接注入し、中枢神経系の髄鞘化と神経再生を促す再生医療的アプローチも進められており、重度の発達遅滞を伴う染色体異常の患者に対する将来的な応用が期待されています。
よくある質問(FAQ)
🏥 希少疾患の不安を、ひとりで抱えないために
染色体異常やNIPT陽性で不安が消えないのは、親として当然のことです。
私たちは専門医による正確な医学知識と心の安全を最優先に、ご家族の意思決定に最後まで伴走します。
関連記事
参考文献
- Duplications of 10q – RareChromo.org [外部サイト]
- Trisomy 10p – Orphanet [外部サイト]
- General information about positive NIPT results: Trisomy 10 – Sonic Genetics [外部サイト]
- 10q deletions: breakpoints in 10q25 or 10q26 – RareChromo.org [外部サイト]
- Partial monosomy of chromosome 10 short arms – PMC [外部サイト]
- Severe Hypocalcemia Dependent on Fluconazole in a Newborn With Barakat Syndrome [外部サイト]
- DiGeorge Syndrome – StatPearls – NCBI Bookshelf [外部サイト]
- Distal duplication 10q syndrome – Orphanet [外部サイト]
- 10q26 deletion syndrome – Genetics – Medline Plus [外部サイト]
- The Future of Gene-Editing Treatments for Rare Diseases | Yale School of Medicine [外部サイト]






