脊髄性筋萎縮症(spinal muscular atrophy: SMA)
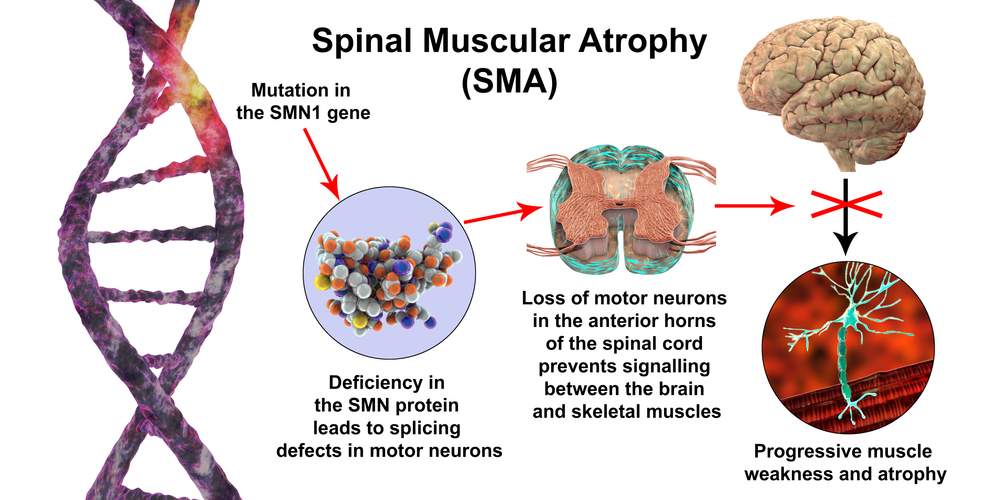
脊髄性筋萎縮症(SMA)は、神経が原因で筋肉に影響を与え、筋肉が次第に弱くなる遺伝性の病気で、常染色体劣性遺伝性疾患では2番目に多い疾患です。脊髄性筋萎縮症(SMA)は、脊髄前角の運動神経細胞の変性が特徴で、左右対称の筋力低下と萎縮を引き起こします。乳児の死亡原因の中で最も多い遺伝子疾患です。主に乳幼児に発症しますが、成人でも発症することがあります。症状や予後はSMAのタイプによって異なります。現在では治療薬もできてきました。
脊髄性筋萎縮症(SMA)とは?
脊髄性筋萎縮症(SMA)は、遺伝性の神経筋疾患で、筋肉が弱くなり、衰えていく病気です。SMAの患者さんは、筋肉の動きを制御する脊髄の特定の種類の神経細胞(運動ニューロン)を失います。この運動ニューロンがないと、筋肉を動かすための神経信号を筋肉に送ることが出来なくなります。萎縮という言葉は、医学用語で小さくなるという意味です。SMAでは、特定の筋肉が使われないために小さくなり、弱くなります。
脊髄性筋萎縮症の確率とは?
米国では、およそ10,000人から25,000人の子供と大人がSMAを患っています。6,000人から10,000人の子供のうち1人が罹患する稀な病気です。日本では、Ⅰ型が2万人に1人、乳児期に発症する脊髄性筋萎縮症(SMA)(Ⅱ型)は約10万人あたり1~2人とされており、欧米より少し少なくなっています。常染色体劣性の遺伝形式をとるので、病的遺伝子の保因者は約70人に1人と推定されます。
どんな人が脊髄性筋萎縮症になるのですか?
SMAの人は運動神経生存タンパクを作るsurvival motor neuron 1;SMN1)遺伝子の欠損または欠損(変異、バリアント)を2コピー受け継いでいます。1つの病的遺伝子は母親から、もう1つは父親から受け継ぎます。SMAの原因となる欠陥遺伝子を1つだけ持っていても、何も症状は現れないので、それを自覚することはありません。
米国人では約50人に1人が、変異したSMN1遺伝子を持っています。日本人ではもう少し少ないとされていますが、Ⅰ型の頻度は2万人に1人なので、約70人に1人という計算になります。これらの保因者は健康なSMN1遺伝子と欠陥や塩基配列に突然変異のあるSMN1遺伝子を1つずつ持っています。保因者はSMAを発症しません。2人の保因者がSMAの子供を持つ確率は4分の1です。
脊髄性筋萎縮症SMAには主に5つのタイプがある
国際SMAコンソーシアムによる分類では、SMAの表現型は、発症年齢と運動発達段階により、いくつかの重症度に分類されています。
- 0型(最重度)
- 0型は胎内で発症します。生命予後は生後約1週と最重症型。SMN2遺伝子のコピー数は1です。
- Ⅰ型(重度)
- ウェルドニッヒ・ホフマン病とも呼ばれ、SMA患者の約60%が1型です。症状は出生時または生後6ヶ月以内に現れます。1型SMAの乳児は、嚥下や吸引が困難です。また、首がすわらない、お座りができないなど、典型的な発達の遅れがみられます。筋肉が弱くなるにつれて、呼吸器感染症や肺の虚脱(気胸)を起こしやすくなります。1型SMAの子どもは、ほとんどが呼吸を支持する治療をしなければ2歳の誕生日を迎える前に死亡します。
IA型はSMN2遺伝子のコピー数は1~2です。胎内で発症し、生後2週間くらいで顕著な症状を示します。
IB型はSMN2遺伝子のコピー数は2~3です。生後3か月くらいで発症し、頸は座りません。
IC型もSMN2遺伝子のコピー数は2~3です。生後3~6か月で発症し、頸は座りますが寝返りやお坐りは出来ません。 - Ⅱ型(中間型)
- 中等症で、臨床経過は様々です。生後6ヶ月から18ヶ月の間に症状が現れます。このタイプは上肢よりも下肢が侵される傾向があります。Ⅱ型SMAの子どもは、座ることはできても歩くことはできません。Ⅱ型SMAのほとんどの子どもは、成人まで生きることができます。殆どの患者さんのSMN2のコピー数は3となっています。
- Ⅲ型(軽度)
- 3型(若年型):Ⅲ型SMA(クーゲルベルト・ウェランダー型、若年性SMAとも呼ばれる)の症状は生後18ヶ月以降に出現します。通常、慢性的な経過をたどります。少なくとも幼児期には自立歩行が可能です。Ⅲ型では成人期まで症状が現れない人もいます。Ⅲ型の症状には、軽度の筋力低下、歩行困難、頻繁な呼吸器感染症が含まれます。時間が経つと、症状は歩いたり立ったりする能力に影響を及ぼすことがあります。Ⅲ型のSMAが寿命を著しく縮めることはありません。SMN2のコピー数は3~4となっています。
- Ⅳ型(成人型)
- 成人型SMAは稀で、通常30代半ばまで現れません。筋力低下の症状はゆっくりと進行するため、Ⅳ型の方のほとんどは、体を動かしながら充実した生活を送ることができます。SMN2のコピー数は4以上となっています。最も軽症で、症例報告は少なく、その有病率も正確にはわかっていません。
脊髄性筋萎縮症の原因
SMAの患者さんは、SMN1遺伝子の一部が欠損しているか、遺伝子が変化して遺伝子産物であるタンパクが機能をうしなっています。健康なSMN1遺伝子はSMN蛋白を産生します。運動ニューロンが生存し、適切に機能するためには、このタンパク質が必要です。SMAの人はSMNタンパク質が十分に作られないので、運動ニューロンが縮んで死んでしまうのです。その結果、脳は頭、首、腕、脚の動き(随意運動)をコントロールすることができなくなります。
脊髄性筋萎縮症は、患者の95%以上で5q13遺伝子座に関連しています。SMN1遺伝子を含むいくつかの遺伝子を含む重要な領域に、逆位や重複が認められます。SMN1のホモ接合体欠失は、これらの症例の98%を占め、乳児期、中間期、成人期の発症が報告されている。SMAの家族における連鎖解析では、患者の2%に大きなde novo欠失が認められます(文献)。de novoは日本語では新生突然変異といって、罹患している人の受精胚を作った卵子や精子で遺伝子の塩基配列の変化が起こってしまって病的遺伝子ができたことを意味しています。
SMN2はセントロメア重複領域にあるSMN1遺伝子と相同性が高い(塩基配列が似ている)遺伝子です。セントロメア遺伝子のエクソン7におけるヌクレオチド置換(C→T)は、コードされるアミノ酸を変えることはないのですが、エクソン7をスキップ(読み飛ばし)させてしまいます。SMN1遺伝子全体のホモ接合性欠失は、ほとんどの患者において脊髄性筋萎縮症SMAの原因となるのですが、SMN1とSMN2を含むハイブリッド遺伝子が報告されています(文献)。したがって、SMN1エクソン7の欠失は脊髄性筋萎縮症SMAの分子診断に用いられています。ホモ接合性のSMN2欠失は正常対照者の5-9%に認められ、病的なものではないと考えられています(文献)。
SMN2遺伝子を持っている人は、少量のSMNタンパク質を産生することができます。SMN2遺伝子ではエクソン7の6番目のヌクレオチドがC(シトシン)からT(チミン)に置き換わっているため、スプライシングの過程でほとんどのエクソン7がスキップされ、産生されるSMNタンパク質の約90%が短縮型の非機能性のものとなります。その結果、SMA患者における機能性SMNタンパク質は、SMN2遺伝子から産生されるわずかな量(10%と言われている)のみとなります。SMN2遺伝子は最大8コピーあります。通常ならばSMN1遺伝子が十分な量のSMNタンパクを作るためと考えられていますが、SMN2遺伝子の産物であるSMNタンパクは体内ではすぐに分解されます。SMN2遺伝子が複数あると、SMN1タンパク質の欠損を補うことができるため、SMN2遺伝子のコピー数はSMAの重症度に影響を与えます。まれに、SMN遺伝子以外の変異(5番染色体以外)がSMAの原因となることがあります。
脊髄性筋萎縮症の症状はどのようなものですか?
SMAの症状は、タイプによって異なります。一般的に、SMAの人は、筋肉を動かす力や強さが徐々に失われていきます。筋力低下は年齢とともに悪化します。この病気は、胴体や首に近い筋肉に深刻な影響を与える傾向があります。SMAの患者さんの中には、歩いたり、座ったり、立ったりすることができない人もいます。また、これらの動作が徐々にできなくなる人もいますし、うまれた時からある人もいます。
脊髄性筋萎縮症はどのように診断されますか?
SMAの症状の一部は、筋ジストロフィーなどの神経筋疾患から生じる症状と似ています。症状の原因を見つけるために、医療従事者は身体検査と病歴聴取を行います。また、SMAの診断のために、次のような検査を行われます。
- 血液検査
- クレアチンキナーゼCKの濃度を調べます。筋肉に問題があると、この酵素が血流に放出されます。
- 遺伝子検査
- SMN1遺伝子に問題があるかどうかを調べます。遺伝子検査では95%の患者さんでSMN1遺伝子の変化が見つかります。
- 神経伝導検査
- 筋電図(EMG)により、神経の筋肉や神経の電気的活動を測定します。
- 筋生検
- まれに筋生検が行われます。生検では、筋肉の萎縮や減少が見られることがあります。
脊髄性筋萎縮症は妊娠中に診断されるのでしょうか?
SMAの家族歴がある場合、出生前検査で発育中の胎児がこの病気であるかどうかを判断することができます。これらの検査は、流産や妊娠の喪失のリスクをわずかに増加させます。SMAの出生前検査には羊水検査や絨毛検査が行われますが日本では倫理的観点から実施しているところを探すことが難しいかもしれません。
最近ではNIPTでお子さんに脊髄性筋萎縮症があるかどうかを調べることが出来るようになりました。NIPTによる脊髄性筋萎縮症の検査は妊娠10週以降に行われますが、両親が同じ病的バリアントを持っている場合には検査が難しくなりますので、NIPTで脊髄性筋萎縮症を検査したい場合には、その前にパパとママのSMN1遺伝子検査をしておく必要があります。ミネルバクリニックではこの検査をお取り扱いしておりますので、検査をご希望になる方は早めにご相談を下さい。
脊髄性筋萎縮症はどのように管理・治療するのですか?
SMAを治す方法はありません。治療法は、SMAの種類と症状によって異なります。多くのSMA患者さんは、理学療法や作業療法、装具、松葉杖、歩行器、車いすなどの補助具を使用することで効果を得ています。また、疾患修飾療法が有効な場合もあります。疾患修飾療法とは、SMNタンパク質の産生を促進する薬剤です。
疾患修飾療法
運動神経を保護するSMNタンパク質の産生を促進する薬剤です。
- ヌシネルセン(スピンラザ)
- この薬は脊柱管周囲の空間(髄膜腔)に注射します。SMN2遺伝子ではエクソン7の6番目のヌクレオチドがシトシンCからチミンTに一塩基置換されています。このため、スプライシングの過程でほとんどのエクソン7がスキップされ、産生されるSMNタンパク質の90%が短縮型のものとなり、タンパクとしての機能を有していません(文献)。残り10%は完全長のSMNタンパクが産生されます。エクソン7とエクソン8の間のイントロンにアンチセンスであるヌシネルセンを結合させることで、エクソン7の読み飛ばしを防いで完全長のSMNタンパクの産生を増やし、運動神経細胞を支持する効果が確認されました。数か月おきに定期的に治療を繰り返すことが必要です。ヌシネルセンの投与にはSMN2遺伝子のコピー数が2以上あることが必要となります。
- リスジプラム(Evrysdi)
- 大人と2ヶ月以上の子供に効果があります。リスジプラムは、毎日経口服用します。SMN2 プレmRNAの選択的スプライシングを修飾し、エクソン7が欠損したΔ7 mRNAからエクソン7を含んだ完全長mRNAへと産物を変えることで、機能性SMNタンパク質の産生量を増加させると考えられます。
遺伝子治療
オナセムノゲンアベパルボベクゾル(商品名:ゾルゲンスマ)は、人工的に作成したSMN遺伝子を外部から患者さんの体内に導入することで、患者自身が半永久的にSMNタンパクを作ることを可能とする治療法です。治療用アデノウイルスベクターという病原性のないウイルスの殻の中にSMN遺伝子を入れて、運動神経細胞に到達するよう設計されています。患者さんにはアデノウイルスベクターAAV9に感染してもらわないといけませんから、成功させるためには2か月程度の期間、免疫抑制状態にするため副腎皮質ステロイドを内服する必要があります。投与時に2歳未満の患者さんだけが投与対象となります。また、アデノウイルスベクターに対する抗体をもっている患者さんには投与ができません。
脊髄性筋萎縮症の合併症
脊髄性筋萎縮症では、時間の経過とともに筋力低下が進行し、筋肉のコントロールができなくなります。可能性のある合併症は以下の通りです。
- 骨折、股関節脱臼、脊柱側弯症(背骨の湾曲)。
- 摂食・嚥下障害による栄養失調と脱水症状(栄養チューブが必要になることもあります)。
- 肺炎や呼吸器感染症。
- 肺が弱く、呼吸に問題があるため、呼吸補助(人工呼吸)が必要な場合がある。
脊髄性筋萎縮症の予防
どうすれば脊髄性筋萎縮症を予防できますか?ということが問題になります。脊髄性筋萎縮症SMAは遺伝性の疾患です。予防するには本人とパートナーの保因者スクリーニング検査をすることが重要になります。SMAの原因となる変異した遺伝子を持っている場合、お子さんがSMAになる可能性や保因者になる可能性について説明することができます。また、両親が双方病的遺伝子を保因していなくても、新生突然変異で揃ってしまうことがあります。新生突然変異de novo mutationがSMAの患者さんの約2%で観察されています。
SMAの家族および親族に対する遺伝カウンセリングにSMAのキャリア検出法が導入されて、再発のリスク評価がより改善されました。両親のキャリアスクリーニング(保因者検査)は、de novo欠失(新生突然変異)が1%に認められることから、出生前診断を勧める前に行うことが望ましいとされています。しかし、両親が同じ染色体上のSMN1重複を持ち、もう一方の染色体上の欠失を伴う場合の有病率は、現在のところ不明です。SMA患者およびその近親者の遺伝子型を正しく解釈するためには、SMN1重複を見分けることも非常に重要となります。
SMAの原因となる病的遺伝子を持っている場合には、お子さんがSMAを発症するリスクを低くするために、着床前遺伝子診断(PGD)を受けることが可能ですが、日本ではいろいろ倫理的に制約があります。
※ミネルバクリニックでは新型出生前診断NIPTによる脊髄性筋萎縮症SMAの検査のお取り扱いを近日中に開始する予定です。
脊髄性筋萎縮症の見通しと予後
脊髄性筋萎縮症の人の予後がどうなのかということが気になります。SMA患者の生活の質と余命は、タイプによって異なります。1型SMAの乳幼児は、通常2歳の誕生日を迎える前に死亡します。2型や3型のSMAの子どもは、症状の重さにもよりますが、充実した人生を送ることができます。大人になってからSMAを発症した人(4型)は、多くの場合、活動的で通常の余命を楽しむことができます。
この記事の著者:仲田洋美医師
医籍登録番号 第371210号
日本内科学会 総合内科専門医 第7900号
日本臨床腫瘍学会 がん薬物療法専門医 第1000001号
臨床遺伝専門医制度委員会認定 臨床遺伝専門医 第755号







