目次
染色体異常は、種々の遺伝的疾患の背景にある主要な要因です。特に、構造的染色体異常は、多様な疾患の発症に深く関与しており、そのメカニズムの理解は医学の領域において重要な意味を持ちます。生殖細胞系の異常は、先天性の遺伝症候群を引き起こすことが知られている一方で、体細胞系の異常は、血液悪性腫瘍などの後天的疾患の原因となることがあります。
臨床的に確認された妊娠のうち、約15%が胎児死亡に終わります。特に、20週未満の胎児死亡(自然流産)では、約50%が細胞遺伝学的異常(染色体の構造や数に起こる変化)が原因であるのに対し、20週以上の胎児死亡(死産)では6~13%のケースとなっています。
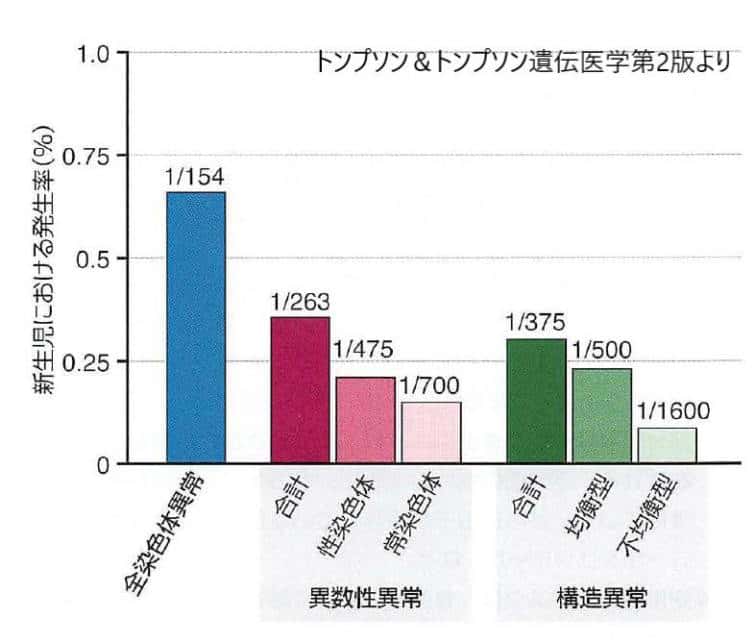
この図は1988年Johns Hopkins大学出版のGenetic disorders and the fetus 第4版に掲載されている内容です。
68000人以上の新生児を染色体検査でスクリーニングした結果、全体としての染色体異常は154人に一人、異数性異常は263人に一人、構造異常が375人に一人となっています。これは1988年のものなので、現在では女性の社会進出による母体の高齢化で、染色体異常は100人に一人と増加しています。
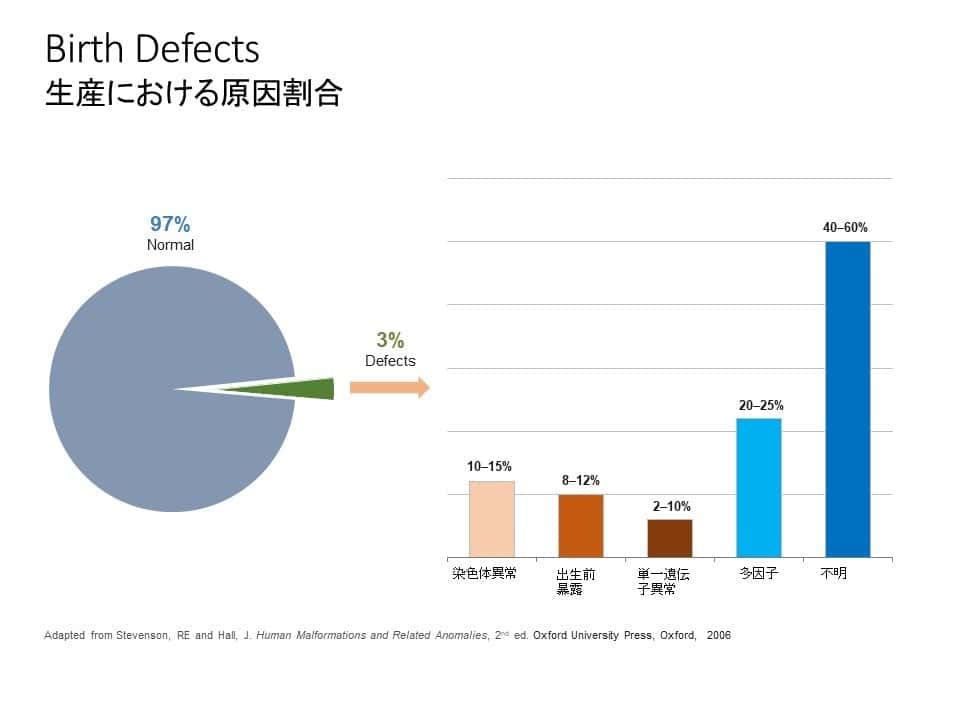
全染色体異常の頻度は生産児(生きて生まれたお子さん)100人あたり1人程度であるため、その影響は臨床医学・社会ともに大変大きいものです。
染色体異常とは
染色体異常は、人間の細胞に存在する染色体の数や構造に異常が生じることで、これにより様々な遺伝的疾患や発達障害を引き起こす可能性があります。染色体はDNAとタンパク質の複合体であり、遺伝情報を保持しています。通常、人間は23対の染色体、合計46本を持っており、これらは親から子へと遺伝情報を伝える重要な役割を果たします。
染色体異常は、その発生する形態に応じて数的異常(異数性)と構造的異常に大別されます。頻度としては、異数性:構造異常=3:2 と概算可能です。以下は、それぞれの異常に関する詳細な解説です。
基本的な定義
染色体異常は大きく分けて二つのカテゴリーに分類されます。数的異常と構造的異常です。
数的異常:染色体の数に異常がある状態で、代表的なものにダウン症(トリソミー21)があります。これは21番染色体が3本存在する状態を指します。
構造的異常:染色体の形状や構造に異常がある状態で、削除、転座、逆位、重複などがあります。これらは染色体の一部が欠けたり、異なる位置に移動したりすることで発生します。
染色体数の異常
正常なヒトの細胞は、2倍体の核型で46本の染色体を持っており、女性は46,XX、男性は46,XYの性染色体を持ちます。数的異常は、通常の2倍体の状態からの逸脱として定義され、次のように分類されます。
●倍数体核型:細胞が1組以上の余分な染色体を持つ状態。例えば、部分胞状奇胎が関連する3倍体状態(69,XXYなど)。
●異数性:特定の染色体が選択的に増加したり減少したりする状態。ダウン症の21番トリソミーは、追加の21番染色体を持つことによって特徴付けられます。染色体の総数が変化すると、ゲノム変異とみなされます。
染色体構造の異常
染色体構造の異常は、染色体の切断によって起こります。染色体の構造が変化する過程を染色体再構成といいます。これには、次のような変化を含みます。
●均衡型再構成:染色体領域がずれるものの、遺伝物質の損失や重複が伴わない状態。しばしば遺伝され、表現型異常とはあまり関連しません。ただし、遺伝子を直接破壊するブレークポイントや、性染色体と常染色体間の物質の置換によって例外があります。
●不均衡な再構成:部分的なトリソミーやモノソミーを引き起こし、先天異常や発達遅滞の原因となることが多い。
●微視的なレベルでの変異:核型分析では検出できないほど小さい変異は、DNA分析によって検出されます。これには次のような遺伝子変異が含まれます。
・一塩基対置換:一つのヌクレオチドが他のものに置換される。
・挿入と欠失:DNA配列に余分なヌクレオチドが加わるか、または失われる。
・重複:あるDNA配列が複製され、同じ遺伝子のコピーが増える。
・ヌクレオチド反復の拡大:特定のヌクレオチド配列が繰り返し拡大される。
・逆位:染色体上のある断片が逆転する。
これらの微視的な変異は、単一のヌクレオチドの変更から数メガベースにわたる大規模な配列の変化までと幅広く、それぞれが異なる遺伝的病態を生じさせる可能性があります。
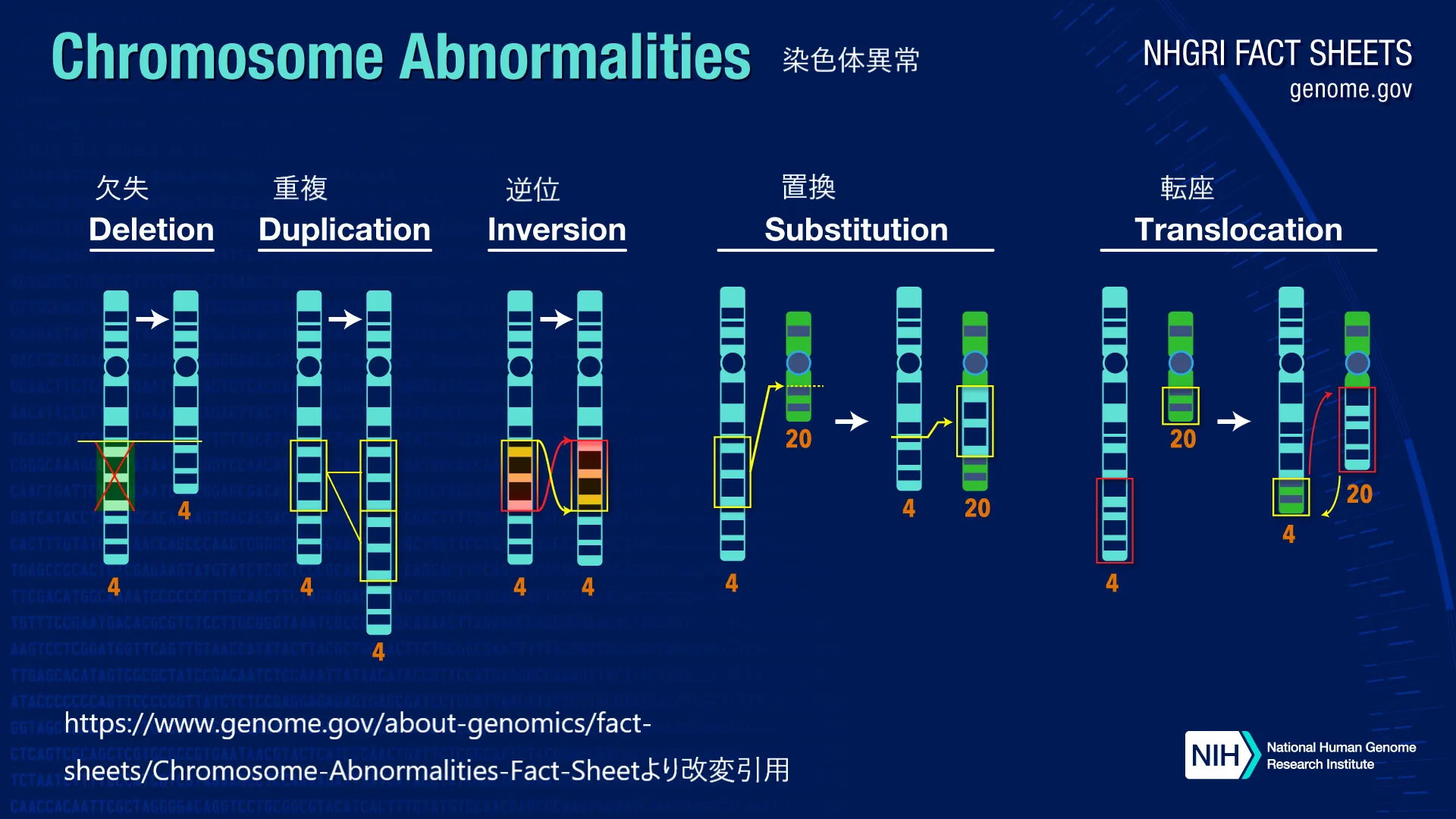
染色体構造異常:染色体切断のメカニズム
染色体の切断について分かりやすく説明します。
染色体の切断は、DNAの二本鎖が内部や外部の原因で切れることによって起こります。内部の原因での切断は、DNAの複製や修理、組み換え、転写などの過程で一時的に必要とされるものです。また、細胞の老化や損傷、細胞死を調節するために恒久的な切断が起こることもあります。外部からの原因としては、放射線の照射や特定の化学物質への暴露などがあり、これらによってDNAが損傷を受けます。
DNAが切れた場合、損傷した部分を取り除き、新しい正常なDNAの配列で修復することができます。この修復過程は高い精度で行われるのですが、DNAの二本鎖が両方とも損傷した場合、修復は複雑になり、エラーが起こりやすくなります。このようなエラーによる変化は、遺伝子の構造や機能に影響を与え、がんのような病気を引き起こす可能性があります。
さらに、染色体の切断は染色体の再配列にも関わります。これには染色体の位置の入れ替わりや、一部の欠失、逆位などが含まれます。
転座
転座には均衡型転座を持つ多くの人が表現型的には正常であるとされていますが、初期の研究では施設入所者の中で均衡型転座の発生率が顕著に高いことが指摘されました。この現象は、遺伝子が直接損傷を受けるか、染色体のごく小さな部分が失われる「位置効果」によると考えられています。
染色体転座には、相互転座、ロバートソン転座、挿入転座(非相互転座)の3つの主なタイプがあります。
相互転座では、2本の非相同染色体が切断され、分離した部分が入れ替わります。これは先天性で発生率は0.2%とされ、まれに2本以上の染色体が関与することもあります。通常、相互転座では遺伝物質の損失はありませんが、減数分裂時には正常染色体と転座染色体が4価構造を形成し、異常な配偶子が生じることがあります。理論的には産生される配偶子の50%が異常であると予想されますが、実際には異常な生殖子を持つリスクが15%を超えることはまれです。
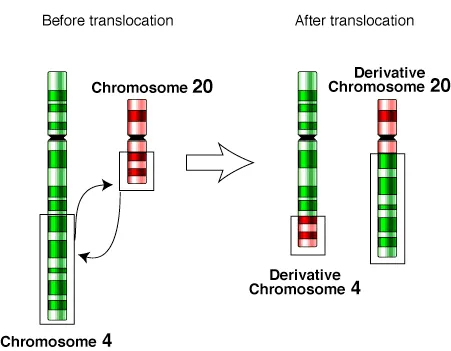
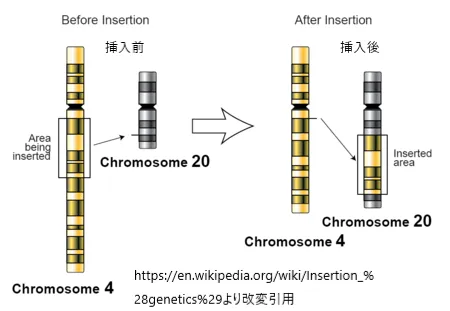
上の図は、第4染色体と第20染色体の相互転座で第4・第20派生染色体が生じる過程を示しています。
相互転座は、血液悪性腫瘍やその他の新生物においても体細胞突然変異としてよく見られます。1つの細胞における相互転座は、2つの遺伝子を隣接させ、そのうちの1つをアップレギュレーションするか、新しい融合遺伝子を作ることでがん遺伝子を活性化します。このプロセスは、影響を受けた細胞のクローン性増殖を引き起こし、直接腫瘍形成につながります。
例えば、t(14;18)転座では、BCL2遺伝子が免疫グロブリン重鎖遺伝子と隣接し、BCL2の過剰発現を引き起こします。これは濾胞性B細胞リンパ腫患者の約80%に見られる転座です。また、t(9;22)転座は、BCR遺伝子とABL1遺伝子の融合を引き起こし、慢性骨髄性白血病の患者に存在します。
これらの転座は、細胞のアポトーシスを防ぎ、細胞周期の制御を直接阻害するなど、様々な機序でがんの発生に寄与します。さらに、発癌性転座に関与する遺伝子は、細胞のシグナル伝達経路を変化させることで、細胞の増殖と生存に影響を与えます。
ロバートソン転座
ロバートソン転座は、特定のヒト染色体(13、14、15、21、22番)で見られる特殊なタイプの転座です。これらの染色体は先中心型で、p-アーム(短腕)が非常に短く、主に染色体サテライトとリボソームRNAの遺伝コードしか含んでいません。そのため、セントロメア(中心体)が染色体の一端に非常に近い位置にあります。ロバートソン転座では、2本の先中心染色体の長腕が結合し、短腕が失われます。これにより、二動的または単動的な融合染色体が形成されることがあります。
このタイプの転座を持つ個体は、46本の染色体が45本に減少しても、表現型には通常影響しません。これは約1000人に1人の割合で見られ、均衡のとれた核型を示します。しかし、ヘテロ接合体の保有者から不均衡な配偶子が生じると、モノソミーまたはトリソミーの胎児が生じる可能性があります。多くのモノソミーおよびトリソミーは致死的で、妊娠初期に自然流産することが多いですが、トリソミー21(ダウン症候群)は生存可能であり、ロバートソン転座を持つ親から生じることがあります。
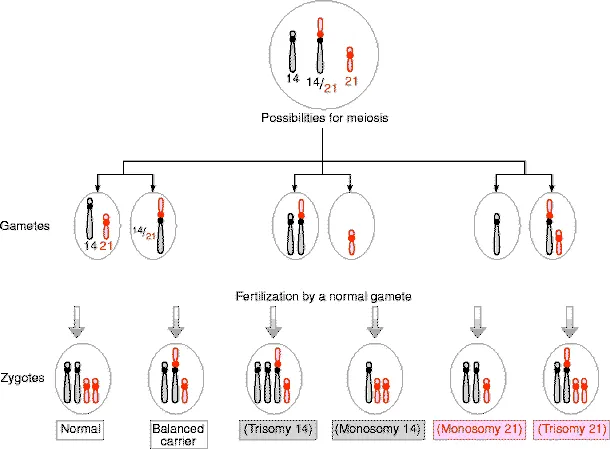
挿入型転座
挿入型転座(IT)は、相互転座とは異なり、「ドナー」染色体の一部が「レシピエント」染色体に組み込まれることによって起こります。このタイプの転座の表現型は、切断点の位置と転座した塩基配列の性質に大きく依存します。アレイ比較ゲノムハイブリダイゼーション(aCGH)技術の進歩により、以前考えられていたよりも挿入型転座が一般的であることが示されており、推定発生率は1/500にもなるとされています。
これらの転座は遺伝的多様性に寄与すると同時に、特定の遺伝性疾患の原因となることがあります。特にロバートソン転座は、ダウン症候群などの特定の症状の発生に関係していることが知られています。
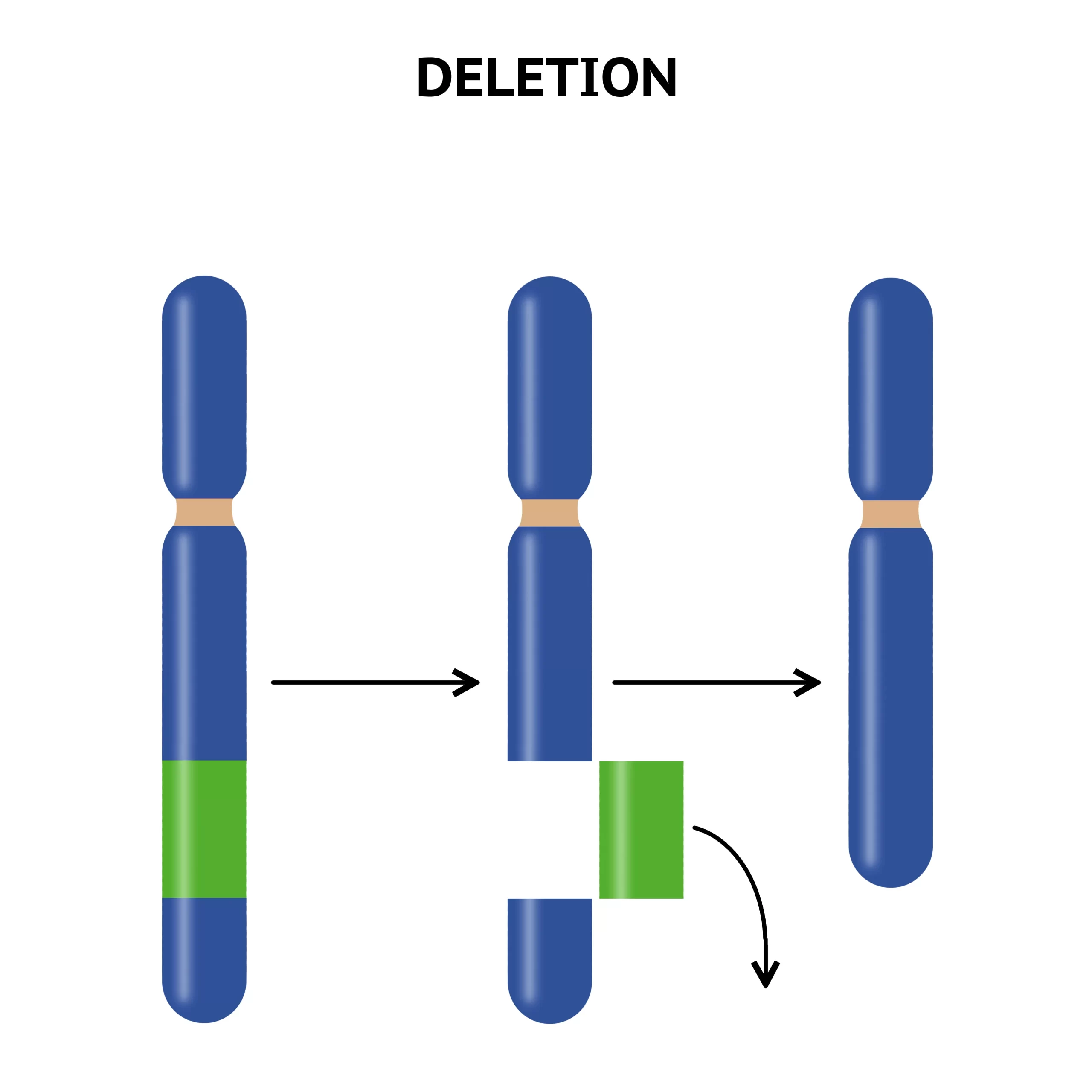
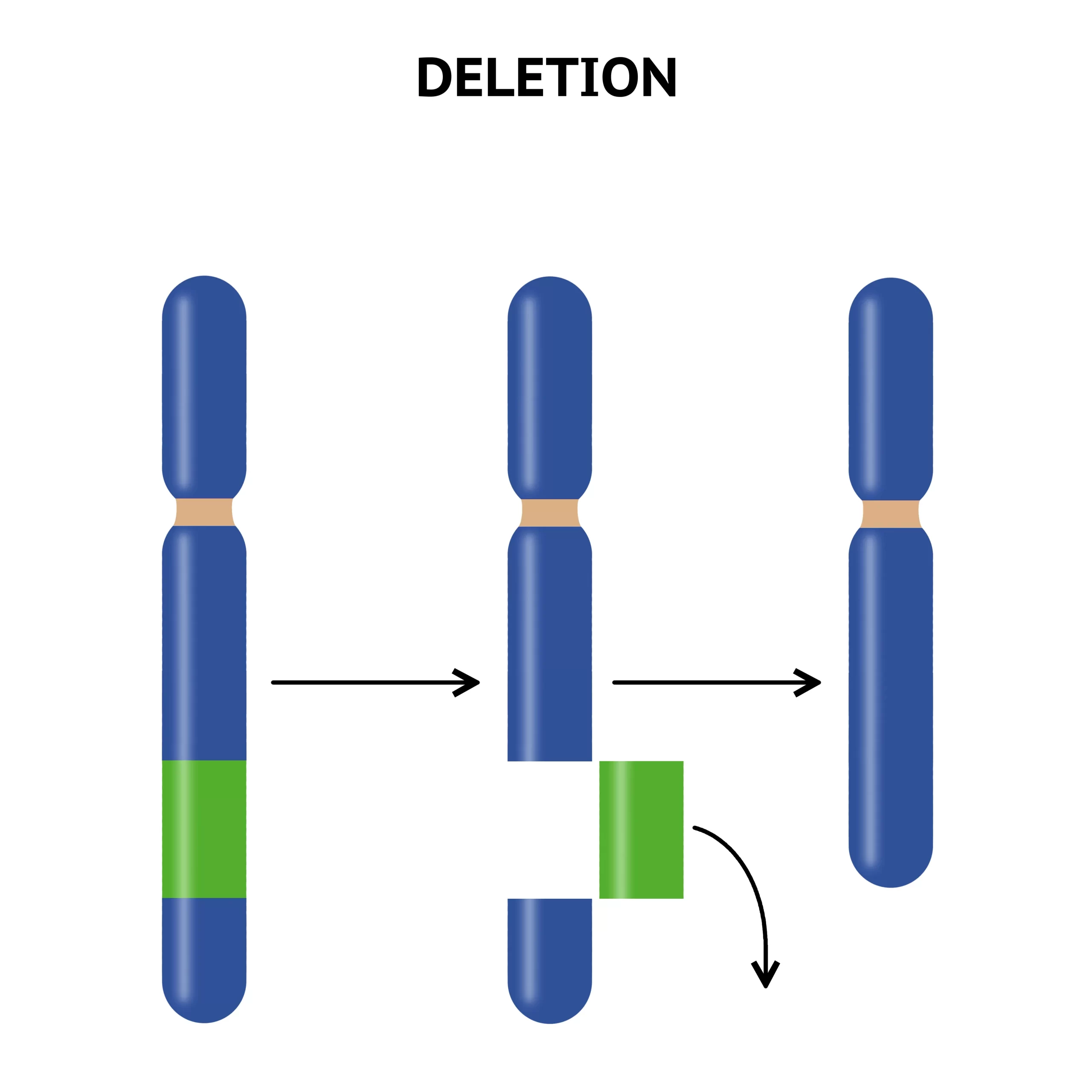
欠失
染色体の欠失は、大きさによってマクロ欠失とミクロ欠失(微小欠失)に分けられ、その検出方法も異なります。マクロ欠失は通常の染色体バンド法で確認できる大きさのもので、3~5メガベース(Mb)以上に及ぶ必要があります。このような欠失は顕微鏡下で直接観察可能で、表現型に影響を与える可能性が高いです。例えば、Cri-du-chat症候群(クリ・デュ・チャット症候群、ネコなき症候群)は5番染色体短腕の末端欠失によって引き起こされ、特有の泣き声や心臓異常など複数の先天異常を特徴とします。
一方、ミクロ欠失は比較的小さな欠失で、高分解能バンディング技術や蛍光in situハイブリダイゼーション(FISH)などのより精密な技術を用いて検出されます。Smith-Magenis症候群やウィリアムズ症候群は、それぞれ特定の染色体領域のミクロ欠失が原因で、特徴的な表現型を示します。
欠失は不等交叉によるものが多く、相同性の高いDNA配列間で誤ったアラインメントが起こり、一方の染色体では塩基配列が重複し、他方では欠失するという現象が生じます。この過程は非同型相同組換え(NAHR)と呼ばれ、多くのミクロ欠失症候群の原因となっています。
これらの欠失は、様々な遺伝的疾患の原因となり得るだけでなく、癌の素因となる場合もあります。たとえば、好酸球増多症候群の患者では、染色体4q12の大きな間質性欠失により新規のFIP1L1-PDGFRA融合タンパク質が生じ、特定の治療薬に反応することが知られています。
染色体の欠失は、疾患の診断や治療戦略の決定において重要な情報を提供し、遺伝学的研究においても重要な役割を果たしています。
逆位
染色体逆位は、染色体の一部が180度回転して元の位置に再統合することを特徴とします。逆位は偏中心性と周中心性の2種類に分けられます。偏中心性逆位は染色体の片方の腕内で起こり、周中心性逆位はセントロメアを挟んで両側の腕が関与します。逆位保因者は通常、外見上は正常ですが、生殖に関しては問題が生じる可能性があります。特に、減数分裂時に逆位染色体が正常染色体とループを形成し、この過程で異常な配偶子が生じることがあります。
●偏中心性逆位pericentromeric
偏中心性逆位は全ての常染色体と性染色体で観察されますが、比較的稀です。この逆位を持つ人は、子孫に異常をもたらすよりも、不妊や反復流産の問題で確認されることが多いです。罹患児のリスクは一般に低いとされていますが、以下のメカニズムにより稀に異常な子孫が生まれることがあります:
伝達された染色体が生命に適合する逆位染色体である場合
逆位領域で組換えが起き、生存不可能な染色体が生じる場合
●周中心性逆位paracentromeric
周中心性逆位では、染色体のバンドパターンと動原体の位置が変わることがあります。逆位により部分的に重複や欠失が発生することがあり、大きな逆位では子孫に表現型異常のリスクが高くなります。減数分裂時に少なくとも1回の交差が必要であり、逆位が大きい場合はその影響を受けやすくなります。
●癌における逆位
体細胞突然変異によって悪性化した細胞では、逆位よりも相互転座や欠失がより一般的に同定されます。逆位は癌患者に見られる染色体異常の中でわずか2%を占め、1本または複数の染色体異常を有するケースで見られます。
染色体逆位は、遺伝的多様性と疾患リスクの理解において重要な役割を果たし、特に生殖に関する問題や癌の研究において注目されています。
染色体構造異常の頻度と影響
染色体構造異常は、欠失、重複、転座、逆位など多様な変化を含み、これらは細胞遺伝学的手法によって検出されます。生児における常染色体欠失の発生率は約7000人に1人とされていますが、常染色体重複の正確な発生率は不明です。これらの構造異常によって引き起こされる症候群は、光学顕微鏡による染色体バンド検査で検出可能なマクロレベルの欠失や重複によるものと、より詳細な分析が必要な微小欠失や微小重複によるものに大別されます。
主な常染色体欠失・重複症候群
●クリ-デュ-シャ症候群 (Cri-du-chat syndrome): 5番染色体の短腕の末端部分の欠失により引き起こされ、特徴的な高い泣き声が特徴です。
●ウィリアムズ症候群 (Williams syndrome): 7番染色体の特定領域の欠失によって引き起こされ、顔貌の特徴、心血管疾患、特異的な性格特性があります。
●ディジョージ症候群 (DiGeorge syndrome): 22番染色体の特定領域の欠失により、免疫系の異常、心臓疾患、顔貌の特徴などが引き起こされます。
これらの症候群は、特定の染色体領域の構造異常により多岐にわたる臨床的特徴を示し、遺伝カウンセリングや疾患の管理において重要な情報を提供します。性染色体の構造異常や微小欠失症候群、微小重複症候群など、他の染色体異常についてはそれぞれ専門的な検討が必要です。染色体構造異常の背後にある機序や影響については、継続的な研究によりさらに理解が深まっています。
染色体の数の異常(異数性)
数型染色体異常、または異数性染色体異常とは、正常な染色体数からのずれ、つまり、通常の46本ではなく、47本以上または45本以下の染色体を持つ状態を指します。このような異常は、1つまたは複数の臓器系に影響を及ぼし、先天奇形を引き起こすことがあります。最も一般的に見られる特徴には、知的障害や低身長があります。さらに、低出生体重、異形(外見の異常)、発育不全もよく見られます。染色体の数の異常は、新生児150人に1人程度で見られる割と多いものです。
異数性染色体異常には、モザイク型が存在する場合があります。モザイク型とは、体内の一部の細胞が正常な染色体数を持ち、他の細胞が異常な数を持っている状態を指します。このモザイクの存在は、個体の生存率や表現型(見た目や症状)にばらつきをもたらします。異数体の中で最も一般的なのは、3倍体、すなわち通常の2倍ではなく3倍の染色体セットを持つ状態です。
21トリソミー(ダウン症)
トリソミー21(ダウン症候群)は出生児中で最も一般的な染色体異常で、出生児730人に1人がこの状態を持っています。ダウン症は主に3つの細胞遺伝学的異常タイプによって引き起こされ、その中で約95%が遊離型トリソミー21(47,+21)、約3~4%が21番染色体のロバートソン転座、1~2%がトリソミー21モザイクです。遊離型トリソミー21は、減数分裂の非分裂エラーによって発生し、余分な21番染色体は大半が母親由来です。ダウン症のリスクは母親の年齢が高くなるにつれて増加しますが、ロバートソン転座によるトリソミー21は母親の年齢とは無関係です。
ロバートソン転座によるトリソミー21の場合、特定の染色体(例えば、14番と21番)が転座し、結果として21番染色体の長腕が3本存在する状態が生じます。このタイプのダウン症は遺伝する可能性があるため、再発リスクが特に重要となります。例えば、バランスの取れたロバートソン転座を持つ親からは、ダウン症の原因となるアンバランス転座を持つ子供が生まれるリスクが存在します。母親が転座保因者の場合、リスクは10~15%と高くなります。
また、21番染色体の長腕の重複はダウン症の稀な型であり、これも異なる形態のトリソミー21をもたらします。この重複はほとんどが新規に発生するもので、DNA多型研究によって、これらの重複染色体の大部分が等染色体であることが示されています。
ダウン症の検出には核型分析や染色体マイクロアレイ(CMA)が用いられますが、これらの方法では異なるタイプのトリソミー21を特定することが可能です。ロバートソン転座によるトリソミー21は、特に遺伝的カウンセリングにおいて重要な意味を持ちます。
18トリソミー
トリソミー18症候群、またはエドワーズ症候群として知られるこの状態は、3つの主な形態があります。
標準的なトリソミー18(47,+18): トリソミー18のケースの約90%は、減数分裂の際の非分裂によって起こります。
18番染色体の転座
モザイク型トリソミー18(47,+18/46)
トリソミー18は、出生児において2番目に一般的な常染色体トリソミーであり、約5500人に1人の出生児に見られます。トリソミー21(ダウン症候群)と同様、母親の年齢が高いほどトリソミー18が発生する可能性が高くなります。罹患児の男女比は3:1です。
トリソミー18の影響は全身のあらゆる臓器に及ぶ可能性があります。主な特徴には以下が含まれます。
子宮内発育制限(IUGR)
筋肉の過緊張
突出した後頭部
小さな口と小顎症
とがった耳
短い胸骨
馬蹄形腎
指の屈曲異常(人差し指が第3指に重なり、第5指が第4指に重なる)
先天性心疾患は罹患者の50%以上に見られ、心室中隔欠損症や動脈管開存症などが一般的です。消化器系の異常は約75%のケースで見られ、メッケル憩室やその他の奇形が主なものです。出生前の診断では、脳梁の異常も比較的よく見られます。
出生前の超音波検査では、IUGRや手の位置異常(”握り手”)、多乳房症などの身体的異常がこの病気の兆候となることがあります。脈絡叢嚢胞も一般的に見られます。
多くのトリソミー18と診断された症例では、子宮内での死亡が多いです。ある調査では、23人のトリソミー18と診断された妊娠のうち、14人が子宮内で死亡し、残りは生後48時間以内に死亡しました。一般に、罹患した乳児の50%が生後2週間以内に死亡し、初年度に生存するのは5~10%です。しかし、学齢期まで生存するケースもあり、1歳以上の生存者では重度の知的障害が見られることが多いです。
13トリソミー
13トリソミー症候群、またはパタウ症候群は、13番染色体の異常によって引き起こされる遺伝的障害です。この症候群は、身体的特徴、発達遅延、およびしばしば重篤な健康問題を特徴とします。13トリソミー症候群には、主に以下の3つの異なる原因があります。
1. 13トリソミー(47,+13)
この状態は、体細胞の各細胞に13番染色体が3つ存在することによって生じます。これは減数分裂時のエラーが原因で、特に母体の年齢が高い場合に発生しやすいとされています。この型はパタウ症候群の最も一般的な形態です。
2. 不均衡なロバートソン転座
13番染色体の正常なコピーが2つ存在し、さらに13番染色体の長腕の余分なコピーが他の先中心染色体(14、15、21、22番)のいずれかに転座することで、完全な13番トリソミーが発生します。このタイプは母体の年齢とは無関係です。13番と14番染色体間のバランスのとれたロバートソン転座は比較的一般的ですが、早期胚死亡率が非常に高いため、保因者がアンバランスな染色体相補体を持つ子を持つリスクは低いとされています。
3. 13トリソミーモザイク(47,+13/46)
この型では、13番染色体が3つある細胞と通常の2つしかない細胞が混在しています。これは細胞分裂の非分裂エラーによって引き起こされ、母親の年齢とは関係ありません。モザイク型は症状の重さにおいて個体差が大きいことが特徴です。
8トリソミー
トリソミー8症候群、特に完全トリソミー8は全妊娠の0.1%で発生し、通常出生前に致死的です。出生児においてトリソミー8が確認される場合、ほとんどがモザイク状態(47,+8/46)であり、その発生頻度は25,000~50,000人に1人です。この症状には性別の偏りがあり、男性:女性=3:1の割合で見られます。出生前診断では絨毛膜絨毛サンプルにおける限局性胎盤モザイクが一般的で、これは通常胎児には影響しませんが、フォローアップとして羊水穿刺が推奨されます。モザイクトリソミー8の診断は困難であり、特にリンパ球ではなく線維芽細胞培養で異常細胞株が検出されることが多いため、染色体構成が正常であってもトリソミー8モザイクを除外することはできません。
モザイクトリソミー8は表現型に大きなばらつきがあり、脳梁の形成不全や脳室肥大、骨格や関節の異常、先天性心疾患、顔面異形、中等度から重度の知的障害など様々な特徴があります。生後のトリソミー8モザイクは接合後分裂エラーに由来することが多く、親の年齢が通常と上昇しない傾向があります。遺伝カウンセリングは表現型のばらつきにより困難であり、予後、管理、治療は患者の具体的な臨床的状況に応じて異なります。患者とその両親や介護者は悪性腫瘍との関連性についても認識しておく必要があり、知的障害の早期評価と特別支援教育プログラムの提供が推奨されます。
9トリソミー
トリソミー9症候群は、極めて稀で、受胎の約0.1%に影響を与え、多くの場合、出生前に致死的です。完全なトリソミー9の状態では、患者は通常、重度の成長制限、特徴的な顔の特徴(例えば、小顎症、球根鼻、低位耳)、口蓋裂、骨格の異常(例えば、結合部の脱臼)、心臓異常(例えば、心室中隔欠損)、生殖器の低形成、腎臓と脳の異常を抱えています。生後の生存期間は非常に短く、数分から9ヶ月の範囲です。
トリソミー9を持つ生まれた子の多くは、モザイク型(47,+9/46)を示し、羊水細胞や末梢血サンプルで検出されることがあります。しかし、絨毛検査(CVS)で観察されるモザイク型トリソミー9は、胎盤に限局したモザイクである可能性があるため、慎重に解釈する必要があります。トリソミー9モザイクの場合、表現型は完全トリソミー9の生児に見られるものと似ており、発育不全、知的及び運動機能の重度の障害、男性では陰睾、腎嚢胞が一般的です。死亡は通常1歳未満であるものの、より長い生存期間が報告されているケースもあります。
トリソミー9は、減数分裂エラーによって引き起こされるとされ、トリソミーレスキューによって生存胚でモザイクが生じることがあります。二倍体細胞株で一方的親源性二倍体(UPD)が確認された例もあります。この症候群は、他のトリソミー症候群と同様に、母親の高年齢と関連があるとされます。
モザイクの割合と臨床症状の間には相関がなく、生存率の予測には役立ちません。治療法は、異常の有無とその重症度に応じて異なります。異常に対処し、必要な医療や教育支援を提供するため、遺伝カウンセリングおよび早期評価が推奨されます。
その他のまれな常染色体トリソミー
その他のまれな常染色体トリソミーは、ダウン症(トリソミー21)、エドワーズ症候群(トリソミー18)、パタウ症候群(トリソミー13)と比べて頻度が低く、多くの場合、モザイク型で発生します。これらのトリソミーは、特定の臨床的特徴を伴い、症例報告を通じて主に記載されています。
トリソミーの種類と関連する症状
トリソミー7モザイク: 母親の単親性倍数体7(UPD 7)と関連し、シルバー・ラッセル症候群を引き起こすことがあります。この症候群は成長障害や特定の顔貌特徴を特徴とします。
トリソミー14モザイク: 成長障害、知的障害、先天性心疾患(特にファロー四徴症や中隔欠損)、体や顔の非対称性と関連しています。
トリソミー22: モザイクまたはフルトリソミーとして発生することがあり、胎児発育不全(IUGR)、小頭症、耳の異常、頚部の皺、皮膚の弛緩、先天性心疾患、腎異常、長指などが主な臨床的特徴です。非モザイク型トリソミー22は通常、流産に終わり、新生児期早期以降の生存は極めてまれです。
リスクレベルに基づく特定のトリソミックモザイク関連の奇形
2番、16番、22番染色体のトリソミックモザイク: 60%以上の奇形リスク。
5番、9番、14番、15番のトリソミックモザイク: 40~59%の奇形リスク。
12番トリソミーモザイク: 26%の奇形リスク。
7番トリソミーモザイク: 19%以下の奇形リスク。
トリソミー17モザイク: 低リスク。
これらのまれなトリソミーの診断は、羊水穿刺などの出生前診断技術により可能ですが、正確な予後を決定するデータは限られています。診断された場合、遺伝カウンセリングや専門的な医療ケアが推奨されます。これらの症状やトリソミーに関するより詳細な情報やサポートについては、医療提供者や遺伝カウンセリングサービスに相談することが重要です。
三倍体
三倍体症候群は、全妊娠の1~3%に見られ、自然流産の約20%を占める比較的一般的な染色体異常です。しかし、出生児における三倍体症候群の頻度は約10,000人に1人と非常に低く、第3期まで生存する三倍体胎児は通常、重度の胎児発育不全(IUGR)を有しています。
三倍体症候群は、完全に余分な一組のハプロイド染色体(合計69本の染色体)を持つ状態で、この余分な染色体セットは通常、母親または父親からの非分離によるものです。二倍体卵子が受精するか、正常な卵子が二倍体精子と受精すること、または2つの精子による受精(二重受精)によって生じます。このうち、二倍体の精子による受精または二重受精による二倍体の三倍体が最も一般的です(約90%)。
三倍体症候群の表現型は、余分な染色体セットの起源によって異なり、刷り込み効果による2つの異なる表現型があります。胎盤の状態や胎児の成長制限の程度など、三倍体症候群の特徴は、その胎児が二卵性か二倍体かによって異なります。二卵性の三倍体では胎盤が腫大し、部分胞状奇胎に類似した組織像を示すことがあります。一方、二倍体の三倍体では、重度の胎児発育制限や相対的な大頭症が特徴です。
三倍体モザイク症の胎児は、一部が生存可能ですが、完全な三倍体症候群を持つ胎児のほとんどは、子宮内または出生後数ヶ月以内に死亡します。この染色体異常に関連する予後は悲観的であり、遺伝カウンセリングではこの点を考慮に入れる必要があります。
過剰マーカー染色体
過剰マーカー染色体(Supernumerary marker chromosome;SMC)、または追加の構造的に異常な染色体(ESAC)とは、正常な二倍体細胞株に加わる小さく構造的に変化した余分な染色体を指します。従来の細胞遺伝学的手法ではその同定が難しかったものの、蛍光in situハイブリダイゼーション(FISH)や染色体マイクロアレイ(CMA)などの現代の分子技術により、46本の染色体すべてからマーカー染色体を特定できるようになりました。
SMCの発生率は羊水穿刺時および新生児でほぼ同じで、それぞれ1000人当たり0.6~1.5人および0.72人です。143,000の連続した出生前検体に関する研究では、de novo(新規に生じた)SMCの全体的な頻度が0.073%であることが明らかにされました。胎盤組織と胎児組織の核型の違いは、胎盤限局性モザイク(CPM)を反映する可能性があります。
de novoマーカーの場合、表現型異常のリスクは全体で14~30%と推定され、特に15番染色体以外の先端染色体(13, 14, 21, 22番)由来のマーカーでは、表現型異常のリスクが7%と低くなることがあります。高精度の超音波検査で先天異常が検出されない場合、リスクはさらに減少する可能性があります。
SMCの同定と特徴付けには、核型分析、FISH、CMAなどの細胞遺伝学的および分子生物学的手法が用いられます。これらの手法にはそれぞれ技術的な限界がありますが、ゲノムワイドマイクロアレイを適用することにより、マーカーを同定し、ゲノム内の潜在的な異常や不均衡を効率的に検出できます。マーカーの起源と遺伝的構成を正確に特定することは、リスクの計算と遺伝カウンセリングにおいて極めて重要です。
ここでは、一般的なSMC(小染色体マーカー)の4つのタイプについて説明します。SMCは、標準的な染色体数に追加される追加の染色体片として見られます。これらは、染色体の構造的異常によって生じることが多く、特定の遺伝的条件に関連しています。
1. 47,+inv dup(15)
特徴: 15番染色体の近位部の逆重複によって形成される。
サブタイプ: 小さなInv dup(15)(q11)と、PWACRを含む大きなInv dup(15)(q12またはq13)。
頻度: 出生前診断で最も一般的に検出されるSMC。大規模研究では、全マーカーの中でそれぞれ57%と25%を占める。
臨床的影響: 大きなinv dup(15)は知的障害、発達遅延、早期中枢性筋緊張低下、痙攣、自閉的行動などの特徴的な臨床所見と関連している。一方、小さいdup(15)の保因者は通常正常な表現型を持つが、まれにUPD 15に起因する症例もある。
2. 47,+i(18p)
特徴: 18番染色体の短腕のテトラソミーを引き起こす等染色体。
頻度: 約625,000人に1人。
臨床的影響: 発育遅延、新生児期の筋緊張低下、小頭症、顔面異形、心臓異常、陰睾など。自閉症の特徴もよくみられる。
3. 47,+i(12p)
別名: Pallister-Killian症候群。
特徴: 12番染色体の短腕のテトラソミーを引き起こすアイソクロモソーム。正常細胞株とのモザイクが頻繁に起こる。
臨床的影響: Pallister-Killian症候群に関連している。
これらのSMCは、特定の遺伝子領域の異常な重複または構造的変化によって形成され、発達遅延、知的障害、および他の多くの臨床的特徴を伴う可能性があります。特定のSMCの存在は、特定の遺伝的状態の診断に重要であり、適切な遺伝カウンセリングおよび管理戦略の提供に役立ちます。
Pallister-Killian症候群とCat Eye Syndrome(キャッツアイ症候群)
Pallister-Killian Syndrome(パリスター・キリアン症候群)とCat Eye Syndrome(キャッツアイ症候群)は、両方とも珍しい染色体異常によって引き起こされる遺伝症候群ですが、それぞれに特有の原因と臨床的特徴があります。
●Pallister-Killian Syndrome(パリスター・キリアン症候群)
特徴
重度の知的障害、痙攣、筋緊張の低下とそれに続く拘縮
粗く異形な顔貌、大きく異常な耳、短い首、まばらな毛髪
出生前には多乳房症と先天性横隔膜ヘルニアが超音波で観察されることが多い
診断
染色体分析(核型、FISH、CMA)が推奨される
47,+i(12p)の細胞はモザイク症例であり、皮膚線維芽細胞で検出されやすい
皮膚生検や頬粘膜塗抹標本など異なる組織の検査が推奨される
管理
早期評価と必要に応じた外科的介入
発達遅滞が軽度の場合は、特別支援教育プログラムの検討
●Cat Eye Syndrome(キャッツアイ症候群)
特徴
耳介前タグおよび/またはピット、虹彩コロボーマ
先天性心疾患(特に全肺静脈還流異常症)、肛門異常、腎奇形、骨格異常、知的障害
臨床症状は軽度から致死的までさまざま
原因
22番染色体の短腕(p)と長腕近位部(q)の逆重複に由来
22qのトリソミーまたはテトラソミーが原因
診断と管理
細胞遺伝学的解析によりinv dup(22)(q11)染色体が検出
両親の染色体解析が推奨される
出生時の心臓、胆道、肛門の異常の評価と早期の介入
これらの症候群は、その珍しさと臨床的特徴の多様性により、過小診断される可能性があります。適切な診断と早期介入は、これらの症候群を持つ個人の生活の質を向上させるために重要です。両症候群とも、専門的な医療チームによる総合的なケアが必要であり、遺伝カウンセリングは患者とその家族にとって非常に価値があります。

ミネルバクリニックでは、「健常なお子さんを抱かせてあげたい」という考えの臨床遺伝専門医の院長のもと、東京都港区青山で、NIPT検査を提供しています。少子化の時代、より健康なお子さんを持ちたいという思いが高まるのは当然のことと考えています。そのため、当院では世界の先進的特許技術に支えられた高精度、かつ、ご希望に合わせてたくさんの疾患検査を提供してくれる確かな技術力のある検査会社を遺伝専門医の目で選りすぐりご提供しています。オンライン診療で全国カバー、採血はお近くの提携医療機関でしていただくことも可能ですので是非ご検討ください。
関連記事:
●NIPTトップページ
●第三世代スーパーNIPT:組織によりDNAメチル化パターンが違うことを利用して正確性を高める世界特許|基本検査/4種類の微小欠失症候群/100種遺伝子の重い劣性遺伝病
●第2世代マルチNIPTカリオセブン:すべての染色体のあらゆる部位を700万塩基のサイズで欠失・重複をチェック/9種類の微小欠失
●第2世代マルチNIPTデノボ:精子の突然変異による重篤な遺伝病44疾患|合計リスクは1/600とダウン症同等に多く、妊娠後期にならないと判らない、ダウン症よりはるかに重篤
●ペアレントコンプリート|母方由来の染色体異数性と父方由来のデノボをセットにしたペアレントの検査
●コンプリートNIPT|第2世代マルチNIPTと第3世代スーパーNIPTジーンプラスをセットにした検査|デノボをセットにしたデノボプラスもございます







