目次
1p36欠失症候群とは?
症状・寿命・原因・診断・治療を臨床遺伝専門医が解説
Q. 1p36欠失症候群とはどのような病気ですか?
A. 1番染色体短腕(1p)の末端領域(1p36)が欠失することで生じる先天性の染色体異常症候群です。
サブテロメア欠失症候群の中で最も頻度が高く、新生児約5,000人に1人の割合で発症します。重度の知的障害・発達遅滞・てんかん・先天性心疾患・特徴的顔貌を主な症状とします。
-
➤
頻度 → 新生児約5,000人に1人(サブテロメア欠失で最多) -
➤
主要症状 → 知的障害(100%)、筋緊張低下(95%)、てんかん(44%)、心疾患(44〜71%) -
➤
遺伝形式 → 約80%が新生突然変異(de novo)、約20%が親の均衡型転座由来 -
➤
診断方法 → 染色体マイクロアレイ検査(CMA)が確定診断のゴールドスタンダード -
➤
寿命・予後 → 適切なケアで成人期まで生存可能(30歳代以上の報告あり)
1. 1p36欠失症候群とは|基本情報
【結論】 1p36欠失症候群は、1番染色体短腕(p)の末端領域である1p36が欠失することで生じる先天性の染色体異常症候群です。サブテロメア(染色体末端部)欠失症候群の中で最も頻度が高く、厚生労働省の指定難病197番に認定されています。
「お子さんの発達が気になる」「検査で1p36欠失が見つかった」という方は、この病気について正確な情報を知ることが大切です。本症候群は多彩な症状を示しますが、欠失領域の大きさや含まれる遺伝子によって症状の程度には個人差があります。
💡 用語解説:「サブテロメア欠失」とは?
染色体の末端部分を「テロメア」と呼び、その内側の領域を「サブテロメア」といいます。サブテロメア領域は遺伝子が密集しているため、この部分が欠失すると複数の遺伝子が失われ、多彩な症状を引き起こすことがあります。1p36欠失症候群はその代表例です。
1p36欠失症候群の概要

出典:Unique (RareChromo.org)
| 項目 | 内容 |
|---|---|
| 疾患名 | 1p36欠失症候群(Monosomy 1p36 Syndrome) |
| 別名 | モノソミー1p36、1p36微小欠失症候群 |
| OMIM | #607872 |
| 指定難病 | 197番(厚生労働省指定難病) |
| 頻度 | 新生児約5,000〜10,000人に1人 |
| 性差 | 女性に多い傾向(約65〜75%が女性) |
| 遺伝形式 | 約80%が新生突然変異(de novo)、約20%が親の均衡型転座由来 |
⚠️ 知っておきたいポイント
1p36欠失症候群は「隣接遺伝子症候群」の一種です。欠失領域には脳・心臓・骨格の発達に関与する複数の重要遺伝子が含まれており、これらが同時に失われることで多彩な症状が現れます。欠失の大きさは患者さんによって異なり、一般に欠失領域が大きいほど症状が重い傾向がありますが、必ずしも単純な相関ではありません。
欠失のパターン
1p36欠失症候群における染色体欠失のパターンは主に3種類に分類されます。
末端欠失(約50%)
- •
染色体末端(テロメア部位)を含む欠失
- •
最も一般的なパターン
- •
単一の切断点から末端まで欠失
中間欠失(約30%)
- •
末端に近い内部領域の欠失
- •
テロメア領域は保持される
- •
2か所で切断が発生
複合的再編成(約20%)
- •
複雑な染色体転座・逆位を伴う
- •
他の染色体部分の重複を伴うことも
- •
親の均衡型転座由来が多い
2. 1p36欠失症候群の主な症状
【結論】 本症候群の症状は知的障害(100%)、筋緊張低下(95%)、てんかん(44%)、心疾患(44〜71%)、特徴的顔貌など多岐にわたります。症状の現れ方には個人差が大きく、欠失領域の大きさや含まれる遺伝子によって多様な表現型を示します。
症状の出現頻度
Battagliaらによる60例の大規模調査およびUnique患者会データに基づく症状頻度です。
| 症状カテゴリー | 頻度 | 詳細 |
|---|---|---|
| 知的障害・発達遅滞 | 100% | 中等度〜重度が大半。運動・言語発達の両面で遅れ |
| 筋緊張低下 | 95% | 新生児期から顕著。哺乳困難の原因にも |
| 言語発達遅滞 | >90% | 多くは話せないか数語のみ。理解力は表出より良好な傾向 |
| 脳構造異常 | 88% | 脳室拡大、水頭症、脳梁低形成、小脳低形成など |
| 心臓の異常 | 44〜71% | 心筋症(約30%)、心室中隔欠損、動脈管開存など |
| 視覚の異常 | 約50% | 斜視、屈折異常、視神経低形成、網膜異常 |
| 行動上の問題 | 約47% | 自傷行為、かんしゃく、他害行為 |
| てんかん | 約44% | 点頭てんかん、Lennox-Gastaut症候群など難治性も |
| 聴覚の異常 | 約28% | 伝音難聴・感音難聴いずれも報告 |
特徴的な顔貌
1p36欠失症候群には比較的一貫した特徴的顔貌が認められ、専門家にとって診断の手がかりとなります。
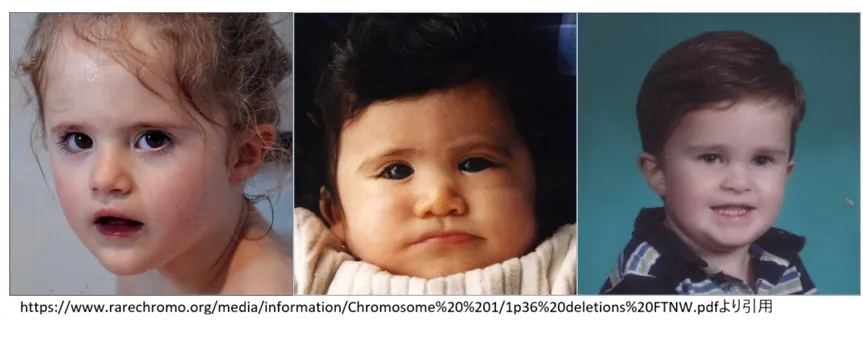
出典:Unique (RareChromo.org) より引用
-
•
眉毛:直線的で平坦な眉
-
•
眼:眼窩が深く目が奥まっている(deep-set eyes)、眼裂がやや短い
-
•
鼻:鼻根部が広く平坦、鼻梁が低い、鼻先が幅広い
-
•
顎:小さく尖った顎(pointed chin)
-
•
耳:低位かつ後方回旋、形態異常
-
•
頭部:前頭突出、大泉門の閉鎖遅延、小頭症傾向(約65%)
心疾患について
心臓の異常は1p36欠失症候群において生命予後を左右する重要な合併症です。
心筋症(約30%)
- •
拡張型心筋症
- •
左室心筋緻密化障害(LVNC)
- •
乳児期から心不全症状を呈することも
- •
PRDM16遺伝子の欠失が関連
構造異常(約40%)
- •
心室中隔欠損(VSD)
- •
心房中隔欠損(ASD)
- •
動脈管開存(PDA)
- •
軽度なら自然閉鎖や経過観察のみも
⚠️ 最新研究(2023年):米国ユタ大学の研究で、PRDM16遺伝子が欠失している群の38%で心筋症を発症(PRDM16が残存している群では8%)と報告されました。特に女性患者で心筋症リスクが高いことも示されています。

🩺 院長コラム【症状の個人差について】
1p36欠失症候群の診断を受けたご家族が最も知りたいのは、「うちの子はどの程度の症状が出るのか」ということだと思います。
正直に申し上げると、欠失の大きさだけで症状の程度を正確に予測することは困難です。同じくらいの大きさの欠失でも、含まれる遺伝子の組み合わせや、他の遺伝的背景、環境要因によって症状は様々です。
ただし、早期療育と適切な医療介入によって、お子さんの発達ポテンシャルを最大限に引き出すことは可能です。臨床遺伝専門医として、個々の患者さんに合わせた具体的なアドバイスをお伝えしています。
3. 原因と遺伝的背景|責任遺伝子
【結論】 本症候群の原因は、1番染色体短腕末端(1p36領域)に位置する複数の遺伝子のハプロ不全です。主な責任遺伝子としてPRDM16(心筋症)、SKI(顔面・脳発達)、RERE(神経発達・眼)、KCNAB2(てんかん)などが同定されています。
💡 用語解説:「ハプロ不全」とは?
通常、遺伝子は父母から1本ずつ、計2コピー持っています。「ハプロ不全」とは、1コピーが欠失または機能しなくなることで、残り1コピーだけでは正常な機能を維持できない状態を指します。1p36欠失症候群では複数の遺伝子が同時にハプロ不全となるため、多彩な症状が現れます。
主な責任遺伝子と臨床症状
| 遺伝子 | 主な機能 | 関連する臨床症状 |
|---|---|---|
| PRDM16 | 転写共役因子、心臓発達を制御 | 心筋症(拡張型、LVNC)、先天性心疾患 |
| SKI | TGF-βシグナル抑制因子、骨・筋肉・脳発達 | 特徴的顔貌、口蓋裂、知的障害 |
| RERE | 転写共調節因子、胎児期の器官形成 | 神経発達障害、眼の異常、脳形態異常 |
| KCNAB2 | カリウムチャネルβサブユニット、神経興奮性制御 | てんかん、発作感受性増大 |
| MMP23B | 頭蓋縫合の結合に関与 | 大泉門の閉鎖遅延、頭蓋骨形成の遅れ |
| GABRD | GABA-A受容体デルタサブユニット | てんかん、神経発達遅滞 |
欠失の発生機序と遺伝形式
新生突然変異(約80%)
両親は正常で、配偶子形成時または受精卵初期に新たに欠失が発生。家族歴はなく、次子への再発リスクは低い(1%未満)。
親の均衡型転座由来(約20%)
親の片方が均衡型転座を持っており、子に不均衡転座として伝わる。親自身は健康でも、次子への再発リスクが高いため、親の染色体検査が重要。
💡 なぜ女性に多い?
報告された患者の約65〜75%が女性ですが、その理由は明確ではありません。男児の方が胎生期に淘汰されやすい、あるいは女性の方が診断に至りやすいなどの仮説がありますが、結論は出ていません。
4. 1p36欠失症候群の診断方法
【結論】 本症候群の確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のG分染法では検出できない微細な欠失を高精度で検出できます。Battagliaらの研究では、60例中46例がFISHまたはCMAで初めて診断されました。
診断のきっかけ
-
①
新生児期の筋緊張低下・哺乳障害:原因検索として
-
②
原因不明の発達遅滞・知的障害:CMAが第一選択検査
-
③
特徴的顔貌+発達遅滞:顔貌から本症候群を疑って検査
-
④
てんかん+発達遅滞:遺伝学的原因検索として
-
⑤
出生前診断:NIPT陽性例や羊水検査で偶発的に発見
遺伝学的検査の種類
| 検査方法 | 特徴 | 1p36欠失の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | ゴールドスタンダード。数kb〜数Mbの微細CNVを高解像度で検出 | ◎ 検出可能 |
| G分染法(核型分析) | 解像度は5〜10Mb程度。大きな欠失は検出可能 | △ 大きな欠失のみ |
| FISH法 | 特定領域のプローブを使用。迅速な確認に有用 | ◎ 検出可能 |
| エクソーム解析 | 全エクソンをシークエンス。大きな欠失はカバレッジから検出可能 | △ 間接的に検出 |
💡 用語解説:染色体マイクロアレイ(CMA)とは?
CMAは、従来のG分染法では検出できない微細な染色体の重複・欠失(コピー数変異:CNV)を検出する検査です。日本では原因不明の発達遅滞・先天異常に対する保険適用検査として実施されています。
5. 治療と長期管理
【結論】 本症候群には根本的な治療法は存在せず、症状に応じた対症療法・早期療育・継続的支援が中心となります。多職種チームによる包括的アプローチが重要です。
ライフステージ別の管理
| ライフステージ | 主な対応 |
|---|---|
| 新生児期・乳児期 | 哺乳支援(経管栄養も考慮)、心エコー検査、てんかんの早期発見、発達スクリーニング開始 |
| 幼児期(1〜5歳) | 早期療育開始(PT・OT・ST)、視覚・聴覚検査、甲状腺機能チェック |
| 学童期(6〜12歳) | 特別支援教育の検討、行動問題への対応、定期的な心機能評価 |
| 思春期・成人期 | 生活自立支援、就労支援、移行期医療(小児科→成人診療科)、心筋症フォロー |
症状別の治療・対応
発達遅滞・言語障害
- •
早期療育が最も重要
- •
理学療法(PT)・作業療法(OT)
- •
言語聴覚療法(ST)
- •
補助代替コミュニケーション(AAC)の活用
てんかん
- •
発作型に応じた抗てんかん薬
- •
難治性ではケトン食療法も検討
- •
発作コントロールが発達に直結
心疾患
- •
軽度の構造異常は経過観察も可
- •
心筋症にはACE阻害薬・β遮断薬
- •
重度では外科的治療も検討
摂食・栄養
- •
哺乳困難には経管栄養・胃瘻も
- •
胃食道逆流(GERD)の管理
- •
成長後の肥満予防も重要
6. 予後と寿命
【結論】 1p36欠失症候群の生命予後は個人差が大きいものの、適切な医療的ケアにより成人期まで十分生存し得ると考えられています。Unique患者会の報告では30歳代以上の患者さんも確認されています。
生命予後に影響する因子
⚠️ リスク因子
- •
重度の心筋症(心不全悪化リスク)
- •
難治性てんかん(重積発作リスク)
- •
重度の嚥下障害(誤嚥性肺炎リスク)
- •
複合的な重篤合併症
✅ 予後改善因子
- •
早期診断・早期介入
- •
適切なてんかん管理
- •
定期的な心機能フォロー
- •
多職種チームによる包括的ケア
💡 発達の将来像について
Battagliaらの長期経過研究によれば、本症候群の患者さんは年齢とともに常に発達的な進歩を示し続けることが強調されています。発達の速度は遅くとも停滞したままではなく、ゆっくりでも学習・成長していきます。
適切な教育・リハビリにより、文字の読み書きや簡単な計算を習得した成人患者の例も報告されています。多くの患者さんは表情豊かで家族やケア提供者との交流から豊かな情緒を育んでいます。
🩺 院長コラム【寿命・予後についての正直な話】
「寿命」についてお尋ねになるご家族は非常に多いです。正直に申し上げると、現時点で「〇歳まで生きられます」と断言することは不可能です。
ただし、医学の進歩により、かつて致命的だった心疾患も現在では外科治療や内科管理が可能となり、生存率は確実に向上しています。30歳代以上の患者さんの報告もあり、適切なケアを続けることで成人期まで生存する見込みは十分にあります。
大切なのは、「いつまで生きられるか」ではなく、「その時々でお子さんが最大限の発達ポテンシャルを発揮できるようサポートすること」だと私は考えています。
7. 出生前診断について|NIPTと羊水検査
【結論】 1p36欠失症候群は出生前診断で検出可能です。NIPTの微小欠失検査や羊水検査でのCMAで診断されます。ただしNIPTはスクリーニング検査であり、確定診断には羊水検査が必要です。
出生前検査での検出方法
| 検査 | 検出可能性 | 備考 |
|---|---|---|
| NIPT(微小欠失検査) | △ スクリーニング | 偽陽性の可能性あり。陽性時は確定検査が必要 |
| 羊水検査+CMA | ◎ 確定診断 | Gバンド法では検出できない微小欠失を確定診断可能。※学会指針では、原則として超音波での構造異常がある場合などが対象とされています。 |
| 絨毛検査+CMA | ◎ 確定診断 | 妊娠初期(11〜14週)に実施可能 |
遺伝カウンセリングの重要性
出生前診断で1p36欠失が見つかった場合、遺伝カウンセリングで以下のことを丁寧にお伝えします。
-
①
欠失の大きさと症状の関係:一般に欠失が大きいほど重症だが、必ずしも単純な相関ではない
-
②
症状の個人差:同じ欠失でも症状の程度は様々
-
③
両親の検査:親が均衡型転座を持つか確認(再発リスク評価)
-
④
出生後のサポート体制:早期療育、フォロー体制の準備
再発リスク
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも正常(新生突然変異) | 1%未満(生殖細胞モザイクの可能性はあり) |
| 片親が均衡型転座保因者 | 高リスク(転座の種類により異なる、約25%程度のことも) |
8. ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。1p36欠失症候群を含む染色体異常の検査から、結果説明、フォローまで一貫してサポートいたします。
🏥 院内で確定検査まで対応
2025年6月より産婦人科を併設し、羊水検査・絨毛検査も院内で実施可能に。転院の必要がなく、心理的負担を軽減できます。
💰 互助会で費用面も安心
互助会(8,000円)により、陽性時の確定検査(羊水検査)費用を全額カバー。上限なしで安心です。
よくある質問(FAQ)
🏥 一人で悩まないでください
1p36欠失症候群について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
参考文献
- [1] Battaglia A, et al. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121(2):404-410. [PubMed]
- [2] Gajecka M, et al. Monosomy 1p36 deletion syndrome. Am J Med Genet C Semin Med Genet. 2007;145C(4):346-356. [PubMed]
- [3] Heilstedt HA, et al. Physical map of 1p36, placement of breakpoints in monosomy 1p36, and clinical characterization of the syndrome. Am J Hum Genet. 2003;72(5):1200-1212. [PubMed]
- [4] Jordan VK, et al. Genotype-phenotype correlations in individuals with pathogenic PRDM16 variants and 1p36 deletion syndrome: a review. Am J Med Genet A. 2023;191(7):1788-1802. [PubMed]
- [5] Shapira SK, et al. Chromosome 1p36 deletions: the clinical phenotype and molecular characterization of a common newly delineated syndrome. Am J Hum Genet. 1997;61(3):642-650. [PubMed]
- [6] Jordan VK, et al. 1p36 deletion syndrome: an update. Appl Clin Genet. 2015;8:189-200. [PubMed]
- [7] GeneReviews – 1p36 Deletion Syndrome. [GeneReviews]
- [8] OMIM #607872 – Chromosome 1p36 Deletion Syndrome. [OMIM]
- [9] Unique – 1p36 deletions. [Unique]
- [10] 難病情報センター – 1p36欠失症候群(指定難病197). [難病情報センター]







